En los últimos años se han emitido diversas notificaciones de la Cofepris y la FDA sobre el retiro de productos debido a la presencia de dimetilnitrosamina (NDMA, por sus siglas en inglés). La NDMA está clasificada como un probable carcinógeno humano. Este compuesto es un contaminante ambiental que podemos encontrar en algunos alimentos en niveles muy bajos.
El retiro de productos del mercado (o recall) es un procedimiento mediante el cual un lote de fabricación es retirado. Tal procedimiento puede ser por iniciativa propia, o bien, por exigencia de la autoridad sanitaria. Esto es una medida de protección que las empresas realizan en caso de que incurran en una no conformidad, ya que podría representar un riesgo para la salud de los usuarios del producto.
Recall Clase I: situación en la que existe una probabilidad de que el uso del producto pueda causar efectos adversos a la salud o, incluso, la muerte.
Recall Clase II: situación en la cual el uso del producto puede causar efectos adversos reversibles a la salud o cuando la probabilidad de efectos adversos a la salud es remota.
Recall Clase III: situación en la cual el uso del producto no puede causar efectos adversos en la salud.
Retiro del mercado: cuando un producto tiene un riesgo menor que no es sujeto a una acción legal por parte de la FDA. La compañía retira el producto del mercado o corrige la desviación. Por ejemplo, un producto que se retira del mercado debido a la manipulación, sin evidencia de problemas de fabricación o distribución.
Es de gran importancia que exista un procedimiento como parte del Sistema de Calidad de cada compañía sobre las acciones a seguir dependiendo de la etapa dentro de la cadena del suministro en la que la no conformidad se haya detectado.
Las causas de retiro del producto en la industria farmacéutica pueden ser variables y dependiendo de cuál sea dicha causa, el riesgo que implique será mayor. Al detectar la no conformidad, el fabricante debe informar a la autoridad sanitaria la cantidad de producto que ya ha sido distribuida hasta el momento, así como los registros de venta. De esta manera, la autoridad podrá identificar los establecimientos en donde se ha distribuido el producto. Cuando el producto ha sido exportado, se debe hacer llegar al país importador.
Es de gran importancia que se cuente con un sistema para el retiro de productos del mercado de manera oportuna y efectiva en el caso de alertas sanitarias y para aquellos productos de los cuales se tenga el conocimiento o se sospeche que están fuera de especificaciones. Se debe contar con un procedimiento que incluya lo siguiente:
- Nombre del responsable de llevar a cabo y coordinar el retiro del producto.
- Actividades del retiro del producto del mercado.
- Instrucciones de almacenaje del producto retirado.
- Cómo se debe notificar a las autoridades.
- Reporte final que incluya una conciliación entre la cantidad distribuida y la cantidad recuperada.
- Las acciones que deberán tomarse para evitar recurrencia y destrucción del producto.
Se deben revisar los registros de distribución del producto para venta, las muestras médicas o para estudios clínicos que permitan un retiro efectivo del producto y llevar a cabo una evaluación continua del proceso de retiro. Además, se debe evaluar la efectividad del proceso de retiro del producto del mercado mediante simulaciones.
Existe una guía en la FDA para manejar los retiros de un lote del mercado (recalls). En esta guía se menciona que se debe incluir una descripción exacta y completa del producto en cuestión, se debe indicarse lote, fecha de expiración, números de serie, números de catálogo y código de barras (UPC). Se puede agregar una copia de la etiqueta del producto en cuestión para hacer más fácil su identificación a los mayoristas y vendedores al por menor en el retiro del producto.
De acuerdo a la guía de la FDA para recalls, incluyendo el retiro y las correcciones del producto, en la notificación debe identificarse claramente el alcance de este retiro, es decir, si es de venta al por mayor, venta al por menor, o bien, a nivel de usuario.
Las compañías farmacéuticas deben tener dentro de su Sistema de Calidad un Procedimiento para Gestionar el Retiro del Producto del mercado. Es importante que la compañía farmacéutica cuente con un buen sistema que le permita rastrear e identificar tanto las materias primas como el material de empaque que entran en contacto con sus productos terminados. Además, se debe tener bien establecido el destino al que se están enviando sus productos una vez que salen de sus instalaciones. Para realizar el retiro del mercado del producto con la alerta, es importante tener los nombres de los contactos involucrados en el retiro y, de este modo, establecer una comunicación directa lo antes posible. También se debe de notificar a las autoridades sanitarias. Una vez que el producto se recuperó se debe tener un área en el almacén para la colocación del producto retirado.
Es muy importante estar preparados con procedimientos en los cuales se encuentre bien definido cómo actuar en cada caso siguiendo siempre las guías de Buenas Prácticas de Manufactura, así como las Normas vigentes en el territorio.
En julio del 2018, se emitió un comunicado por parte de la FDA sobre varios productos farmacéuticos que contenían valsartán debido a la presencia de la impureza N-nitrosodimetilamina o NDMA, incluso está clasificada por la OMS en el nivel 2A, que equivale a medicamentos con posibles riesgos para las personas. Sin embargo, en ese comunicado se mencionaba que no todos los productos con ese principio activo se iban a retirar del mercado. De acuerdo a la FDA, la presencia de NDMA pudo ser el resultado de cambios en la forma de fabricación de la sustancia.
La Agencia Europea de Medicamentos (EMA, por sus siglas en inglés), al hablar sobre la retirada de este medicamento mencionó que la impureza podría tener su origen en 2012 debido a cambios en el proceso de fabricación del medicamento producidos ese año y que la compañía Zhejiang Huahai Pharmaceuticals (China) informó a los fabricantes europeos. Ese mismo mes, la Cofepris también emitió un comunicado en el que ordenaba la inmovilización y no comercialización de la materia prima y de todos los medicamentos que contenían valsartán de dicho fabricante.
La FDA y la EMA también encontraron trazas de N-Nitrosoetilamina (NDEA) en valsartán producido por Huahai, esta impureza, al igual que NDMA también se encuentra listada como posible carcinogénico. Hay una parte del proceso de manufactura en la que existe el riesgo de formar impurezas genotóxicas. Por décadas, la FDA ha proporcionado directrices y recomendaciones para el control de las impurezas en los principios activos y los fabricantes tienen la responsabilidad continua de testear impurezas en base a la comprensión de su propio proceso de fabricación.
En septiembre de ese mismo año, se encontró NDMA en valsartán de otra compañía, Hetero Labs (India) por lo que los productos realizados con valsartán de esta compañía también fueron retirados.
Para finales de octubre, Ibesartán también se añadió a la lista de medicamentos retirados del mercado. En este producto fabricado por Aurobindo Pharma Limited (India) también se encontraron trazas de NDEA.
En noviembre, un tercer medicamento del grupo de Bloqueadores de los Receptores de Angiotensina II (ARB, por sus siglas en inglés) fue retirado del mercado. En este caso fue losartán, producido por Zhejiang Huahai Pharmaceuticals. En este también se encontró presente NDEA. De este, sólo se retiró un lote de tabletas de losartán potásico/hidroclorotiazida de Sandoz. En ese mismo mes, la FDA publicó una carta de advertencia que envió en noviembre del 2018 a Zhejiang Huahai en la que los criticaba por no haber considerado las implicaciones que tendrían los cambios realizados en 2012 en el proceso de producción de valsartán.
También se encontró NDEA en valsartán producido por la empresa Mylan Pharmaceuticals, que utilizó materia prima de Mylan Laboratories Limited (India). En diciembre, Mylan expandió la retirada de productos.
En febrero del 2019, la EMA publicó los límites temporales de la cantidad de NDEA y NDMA encontrados en estos medicamentos. Después de un periodo de transición, las empresas tendrán que demostrar que sus productos no contienen impurezas cuantificables de NDEA y NDMA antes de poder comercializarlos. La FDA también publicó los límites de estas sustancias.
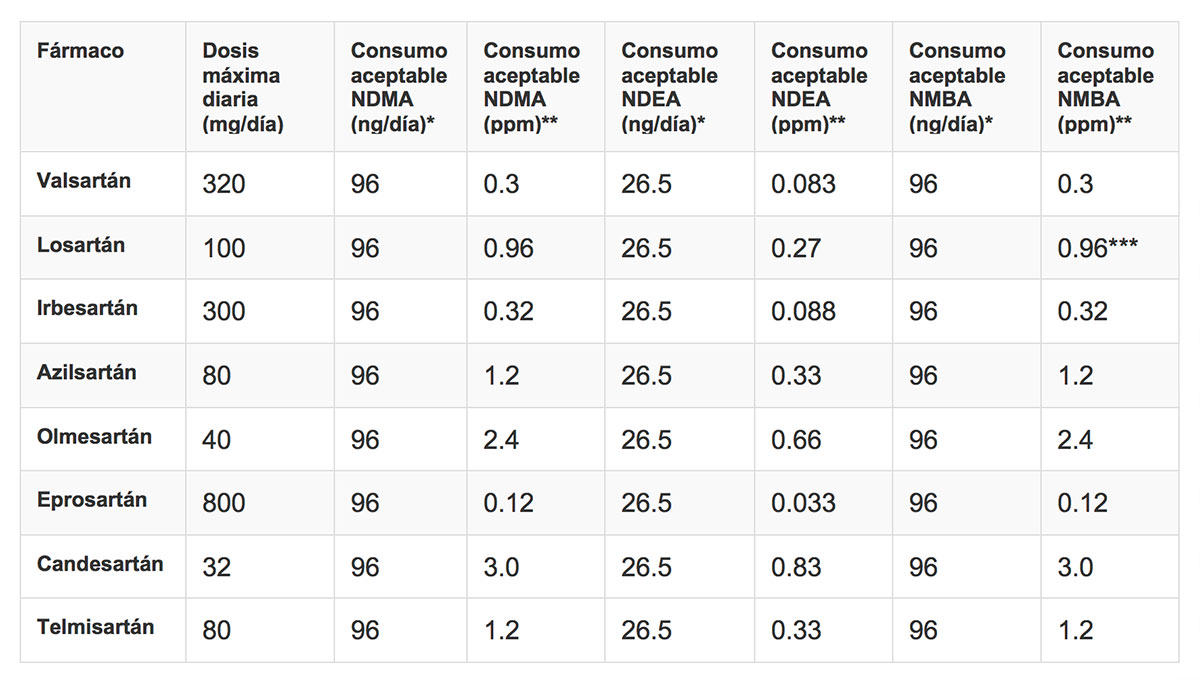
Han continuado retirando algunos productos adicionales. En abril de este año, la FDA dio a conocer nuevas metodologías para detectar e identificar las distintas impurezas de nitrosamina. Dicho método ha sido validado para cuantificar simultáneamente NDMA, NDEA, NEIPA y NDBA en el principio activo valsartán y verificado para medicamentos conteniendo este principio activo. Se deberá verificar para otros activos y medicamentos de esta familia.
Ahora es el turno de la ranitidina que también ha sido muy mencionada en los últimos meses debido a esta misma situación. La ranitidina es un antagonista de la histamina a nivel de los receptores H2 localizados en las células parietales gástricas, que inhibe la secreción de ácido gástrico. Este producto está indicado en el tratamiento de úlcera péptica, gástrica y duodenal, úlcera postoperatoria, esofagitis por reflujo, síndrome de Zollinger-Ellison, prevención y tratamiento de sangrado gastrointestinal superior, y prevención del síndrome de Mendelson.
El comunicado sobre el retiro de la ranitidina fue emitido el 13 de septiembre por la doctora Janet Woodcock, directora del CDER (Centro de Evaluación e Investigación de Medicamentos de la FDA). En este comunicado se notificó sobre la detección de la impureza de nitrosamina: N-nitrosodimetilamina en muestras de ranitidina. Al parecer los niveles que encontró la FDA de NDMA en ranitidina apenas exceden la cantidad de lo que se podría llegar a encontrar en alimentos comunes. La FDA está trabajando con distintas agencias reguladoras y con colaboradores de la industria para evaluar si los niveles bajos de esta impureza pueden implicar un riesgo para los pacientes. Una vez que cuente con esta información, la dará a conocer.
Posteriormente, el 2 de octubre, la FDA mencionó que ha notado que la prueba realizada por un tercero se realiza a temperaturas muy elevadas lo cual genera NDMA en niveles inaceptables. El método de cromatografía de gases publicado previamente por la FDA para la detección de estas impurezas no es adecuado para detectarlas en ranitidina debido a que el calentamiento de la muestra genera NDMA. Debido a la sospecha de la presencia de NDMA, la agencia desarrolló y validó un método de cromatografía líquida acoplado a espectrometría de masas de alta resolución (LC-HRMS, por sus siglas en inglés) para determinar el nivel de NDMA en productos formulados ranitidina.
El 23 de octubre, la FDA liberó un segundo método de cromatografía líquida acoplado a espectrometría de masas de alta resolución con tecnología más al alcance que el método publicado previamente para dar más alternativas a los laboratorios de probar si su producto contiene las impurezas. Sanofi y Dr. Reddy notificaron de su retiro voluntario.
El 28 de octubre, la FDA alerta de otros tres retiros voluntarios de ranitidina, esta vez por parte de Perrigo Company, Novitium Pharma LLC y Lannett Company debido a la presencia de NDMA en sus productos.
En nuestro país, en el comunicado del 5 de octubre, la Cofepris mencionó que estaba evaluando los riesgos a la salud de la población mexicana debido a la posible presencia de dicha sustancia en los medicamentos que contienen ranitidina que son comercializados en nuestro territorio. En ese momento, la Cofepris afirmó no haber encontrado evidencia suficiente para sugerir la suspensión de los tratamientos con este fármaco. Sin embargo, el 29 de octubre, emitió un aviso preventivo en el que indicó a la población en general evitar la compra de medicamentos que contengan ranitidina. Pide a la población que está bajo tratamiento que acuda con su médico para cambiar el medicamento. Al personal médico le pide no prescribir medicamentos con ranitidina. A las farmacias, les pide suspender la comercialización de medicamentos con ranitidina, en tanto se tomen las medidas que garanticen la seguridad del producto.
En nuestro país, la ranitidina es uno de los medicamentos más utilizados para el tratamiento de úlcera gástrica. Son cerca de 42 laboratorios farmacéuticos los que producen medicamentos con ranitidina en México. La medida emitida por la Cofepris es preventiva en lo que se evalúan las diferentes marcas para detectar la presencia de la impureza. Esperemos a que se dé a conocer el resultado final de las pruebas realizadas para determinar si en algún medicamento se encontraron rastros de NDMA. En el caso de que se llegue a encontrar la impureza en algún producto, el medicamento en el que se encuentre será suspendido del mercado mientras los que no contengan la impureza se podrán comercializar de nuevo.
Son varios los países, en los que esta medida preventiva ya fue tomada. Esperemos a ver los resultados de las pruebas que definan qué productos saldrán del mercado y para que tengamos la seguridad de que los productos que consumimos están libres de NDMA.
Es importante que los fabricantes conozcan la posible fuente de formación de NDMA en su proceso de fabricación e incluyan los controles adecuados para reducir la posible formación de estas impurezas carcinogénicas. El nitrito de sodio desencadena una reacción en pH ácido con las aminas, amidas, etc. que pueden estar presentes en los solventes y reactivos y que formarán las nitrosaminas. Por lo anterior, la calidad de los reactivos y solventes utilizados es un factor crítico para asegurar la calidad del producto final.
Aloka Srinivassan, experta de Lachman Consultores, sugiere algunas medidas preventivas para reducir la presencia de estas impurezas:
- Siempre que el fármaco sea una amina secundaria o que alguno de los intermediarios o impurezas en el medicamento sea una amina secundaria, será importante buscar las nitrosaminas correspondientes durante el desarrollo del proceso de manufactura.
- Cuando se utilice nitrito de sodio durante el proceso de manufactura de un fármaco, se deberá realizar una evaluación extensiva de la presencia de nitrosaminas en base a la materia prima usada, reactivos y solventes.
- Cuando se utilicen solventes como DMF, DMA, o DEA en el proceso de manufactura de un fármaco, será prudente buscar presencia de nitrosaminas como NDMA y NDEA.
Si se tiene un buen control del proceso de fabricación, una vez teniendo identificada la fuente de origen de esta impureza carcinogénica se podría llegar a disminuir o, incluso, eliminar la posibilidad de que se formen las nitrosaminas en los productos farmacéuticos.