Recientemente algunos colegas del gremio me han preguntado mi opinión al respecto de la filtración aséptica redundante y quiero enfatizar que este artículo es la opinión técnica de este servidor que documenté, razoné o investigué, afortunadamente no corresponden a un concepto que involucre posición alguna o de algunos, sino a hechos referenciados en literatura y a mi experiencia técnica desde 1981.
Con esto en mente, comenté con algunos de mis colegas, que lo correcto es escribir esta opinión y crear un punto totalmente independiente y esto como un punto de partida de un análisis objetivo con todos los que se quieran involucrar para entender y evaluar la situación de este tema en México y en el mundo (sonó a comercial, pero bueno).
Además, lo tengo que hacer a mi estilo, sólo para sentir la libertad de expresarme y sin el fin de molestar a nadie, solamente ser no ser ortodoxo en un gremio demasiado rígido en momentos.
Primero que nada, definamos filtración redundante, revisemos las bases de la norma de Buenas Prácticas y las prácticas internacionales, hagamos una evaluación de riesgos básicos (NO UN AMEF), esto es una evaluación referencial y los impactos son cuestión de cada usuario de la tecnología, según sea cada proceso y producto, así como permítanme darles mis conclusiones y, por favor, integren las suyas para tener un sustento técnico y evidencia para su sistema de calidad.
¿Qué es un sistema redundante?
En ingeniería, la redundancia es la duplicación de componentes críticos o funciones de un sistema, esto con la intención de aumentar la fiabilidad del sistema, por lo general en forma de una copia de seguridad, o sustitución del componente por fallas o errores, creando un sistema a prueba de fallos.
Se presenta como una solución a los problemas como protección y aseguramiento de confiabilidad del proceso que se realiza. Este tipo de sistemas se encarga de realizar el mismo proceso en más de una estación, ya que si por algún motivo alguna dejara de funcionar o colapsara, inmediatamente otro tendría que ocupar su lugar y realizar las tareas del anterior, es decir, redunda la tarea o la repite de forma funcional, no en valores de operación.
Las técnicas de redundancia han sido usadas por la industria militar, nuclear, petrolera y aeroespacial por muchos años para alcanzar una alta confiabilidad. Una base de datos replicada es un ejemplo de sistema distribuido redundante y es básica en el diseño de sistemas de control lógico programable, conforme a las normas técnicas de la Asociación Americana de Instrumentación (ISA SP840), quien define la redundancia por el tiempo de fallo probable contra unidad de tiempo de uso sin paro, esto no parece ser un modelo de redundancia para filtración aséptica.
Básicamente, la redundancia puede apuntar a dos objetivos:
1.- El aumento de la disponibilidad de un equipo que controla un proceso con otro.
2.- El aumento de la seguridad con que un equipo controla un proceso.
Se desea el aumento de la disponibilidad de un equipo, cuando las detenciones del proceso motivadas por una falla del equipo son costosas, al punto de que con el costo de una o pocas detenciones del proceso se recupera el mayor costo de un equipo redundante.
En otras palabras, en mi opinión el término filtración redundante no es correcto, estamos hablando de una configuración serial, dado que aunque los componentes tienen la misma característica no substituye uno a otro durante la falla de la integridad, dado que el primer filtro no cumple con el criterio de integridad no sería válido seguir la filtración conforme a los criterios de calidad, por ejemplo de la FDA, estos vistos adelante; así mismo, si el segundo filtro falla la prueba de integridad de éste tampoco garantizaría la continuidad el proceso, más bien esta filtración serial es un doble aseguramiento de calidad para mitigar la probable falla del primer filtro en caso de falla de integridad y riesgo de contaminación microbiana, pero esto no es redundancia, lo digo respetuosamente y desde el punto de vista de ingeniería.
Ahora bien, si se entiende filtración aséptica (algunas veces llamada esterilizante), a la técnica de separación física del Biocarga existente en afluente, por medio de una condición de presión, temperatura y propiedades coligativas del afluente al impactar una superficie de material con un poro que permite retener esta carga biológica en la retícula de dicho material. Este mecanismo de separación microbiológica no está reconocido como una operación de esterilización terminal, dado que no aniquila los microrganismos, sino que los separa por retención física, conforme al tamaño de su poro. Este es un debate por separado, lo real es que si el material de filtración o separación no es íntegro, las partículas viables llegarían alegremente al afluente que puede ser producto o cualquier semi-terminado.
 Teniendo esta base, es obvio asegurar que por medio de un doble filtro se mitigará el riesgo de contaminación microbiológica del afluente, pero no se si desde el punto de vista redundante o normativo es válido aprobar un lote con esa condición en México, como si pensáramos, como en los caminos de 18 ruedas, si pones en un rin dos ruedas y se poncha la primera tienes otra en el rin, eso es redundancia y continuidad de funcionalidad.
Teniendo esta base, es obvio asegurar que por medio de un doble filtro se mitigará el riesgo de contaminación microbiológica del afluente, pero no se si desde el punto de vista redundante o normativo es válido aprobar un lote con esa condición en México, como si pensáramos, como en los caminos de 18 ruedas, si pones en un rin dos ruedas y se poncha la primera tienes otra en el rin, eso es redundancia y continuidad de funcionalidad.
Pero en la práctica, los registros actuales de productos estériles por procesamiento aséptico fueron otorgados sin filtración redundante o varios de ellos, así como los estudios de estabilidad fueron realizados para lotes fabricados en su mayoría sin ésta y no se consideró la probable adsorción de activos o el efecto de los lechables o lixiviables (correctamente dicho) en una doble filtración, esto es importante para un sistema legado en México con esta realidad.
Sería interesante conocer las opiniones al respecto de los actores de todos los sectores.
Configuraciones de Filtración Aséptica
Bueno eso es otro tema, pero como son las configuraciones para los sistemas de filtración redundante o serial; en una presentación de Michael Moussourakis para la Sociedad Internacional de Ingeniería Farmacéutica (ISPE) en 2010 sobre los “Tópicos actuales de Filtración Esterilizante” en Tampa, Florida, explica de forma muy sencilla las configuraciones de Filtración Redundante, la cual presento a continuación con los esquemas correspondientes elaborados por su servidor, es claro que los tanques tienen sus sistemas de seguridad, venteos asépticos, drenajes sanitarios y demás.
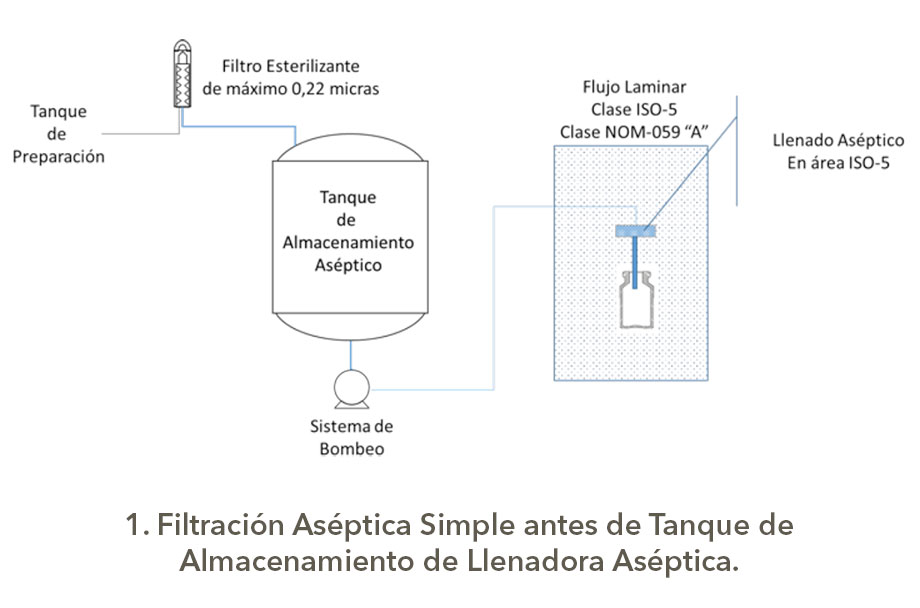
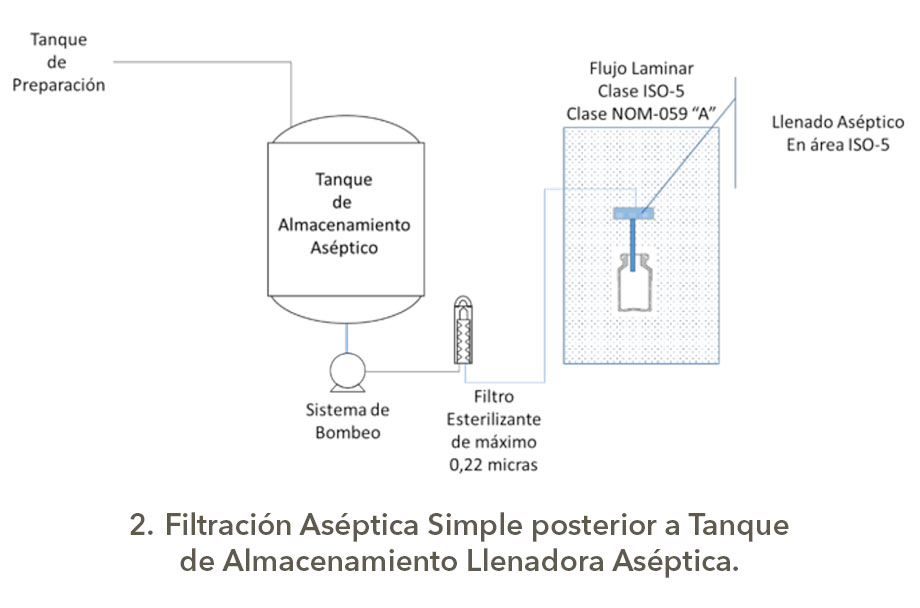
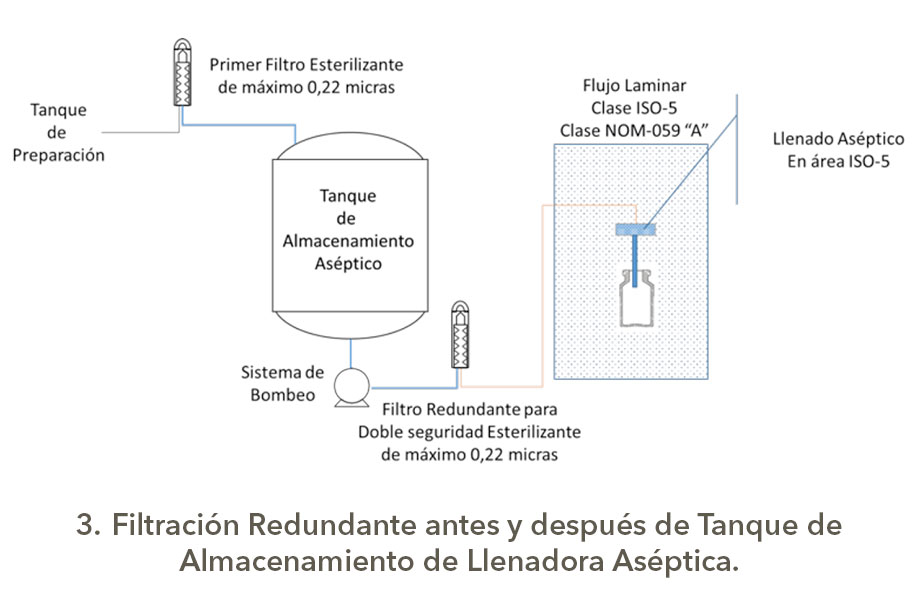
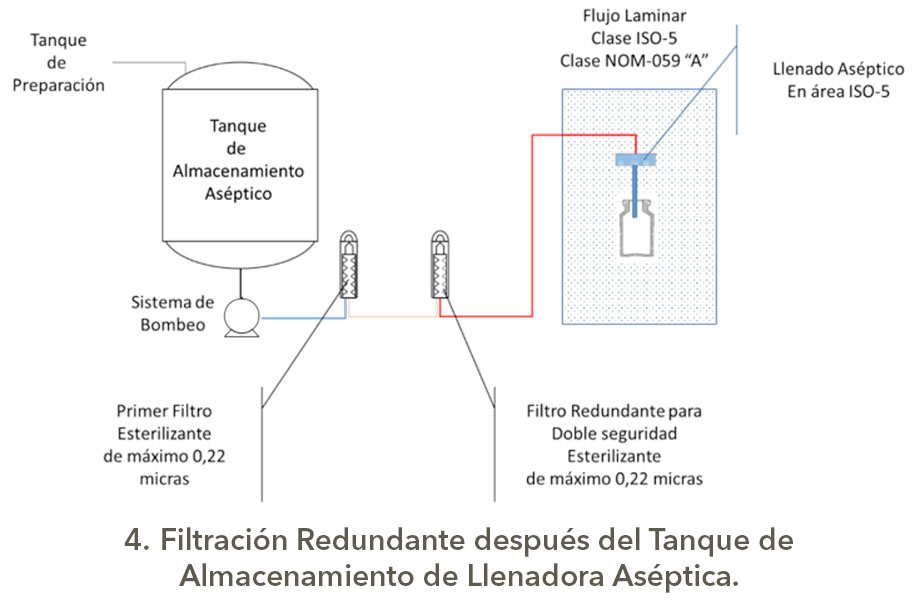
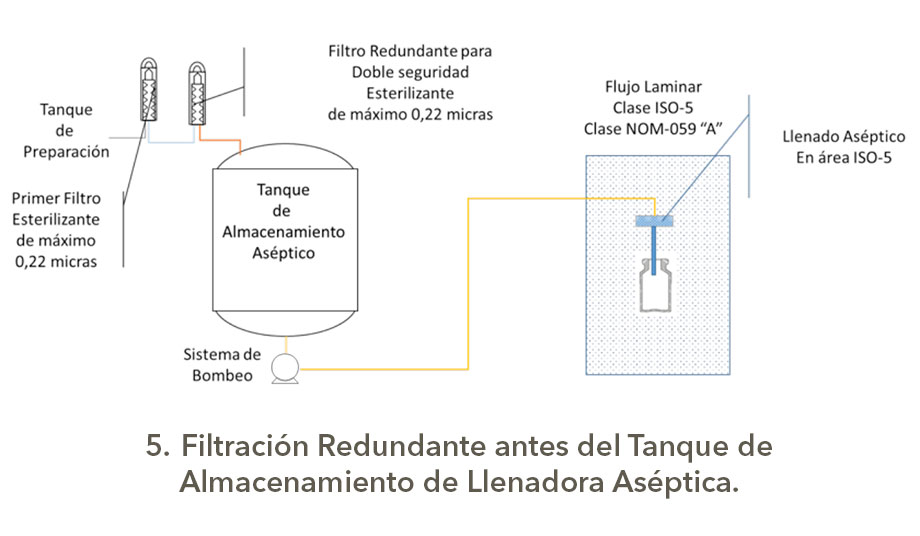
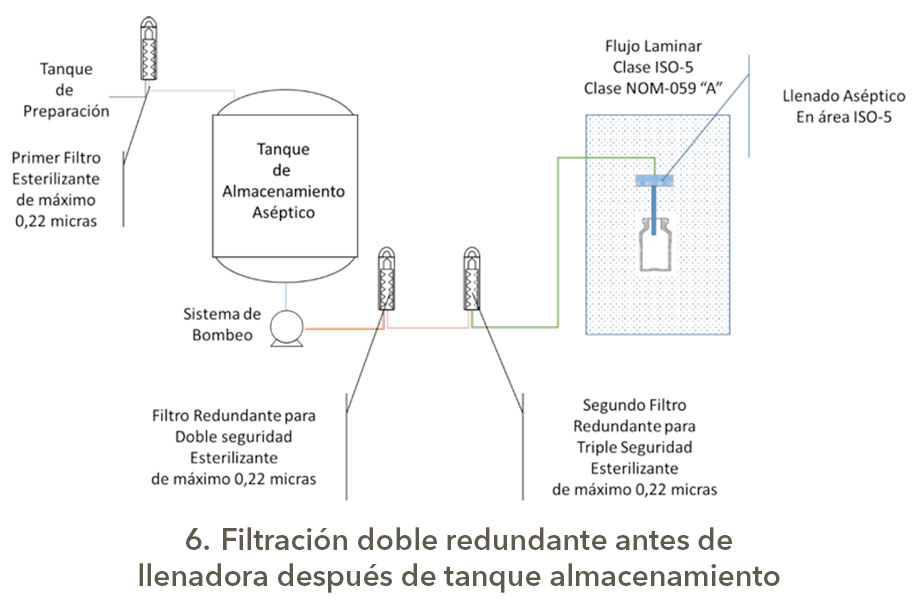
Estas configuraciones para la filtración aséptica tienen diferentes índices de robustez, los cuales dependerán del nivel de carga biológica y origen del granel o afluente a filtrar, así como la criticidad del producto, obviamente no es lo mismo filtrar un genérico con API´s estériles y con excipientes (vehículos correctamente dicho) de Biocarga controlada que productos biotecnológicos o biológicos que son formulados con partículas viables de uso terapéutico o que son por sí mismo promotores de crecimiento, así como si son medios de cultivo usados para manufactura en biotecnología.
En la triada complejidad de proceso, criticidad de producto y nivel de exigencia normativa, radica el punto para la selección del tipo de sistema de filtración con un enfoque en Gestión de Riesgos de Calidad.
¿Qué piensan los líderes?
Las recomendaciones internacionales, como menciona en la presentación del autor anterior, indica que la FDA en la “Guía para la industria de Procesamiento aséptico” vigente 2004 menciona en la sección de eficiencia de filtración sobre este tema.
 Que la “Filtración redundante debe ser considerada en muchos casos”.
Que la “Filtración redundante debe ser considerada en muchos casos”.
El problema es definir “cuáles son muchos casos”, quieren decir ¿que todos los casos de fabricación?
Posteriormente a esto menciona que “cualquiera que sea el filtro o combinación de filtros utilizado, la validación debe incluir retos microbiológicos para simular las condiciones de producción más desfavorables para el material a filtrar y los resultados de los ensayos de integridad de los filtros utilizados para el estudio”.
Esta etapa de validación con los retos debería estar incluida en la Transferencia de Tecnología, pero en nuestra realidad no se incluyó en esta etapa crítica para ser posteriormente incluida en la validación, espero estar equivocado.
Coloco aquí el texto de la guía 2004, para no generar duda en la información con la sección comentada:
“B. Filtration Efficacy
Filtration is a common method of sterilizing drug product solutions. A sterilizing grade filter should be validated to reproducibly remove viable microorganisms from the process stream, producing a sterile effluent.17. Currently, such filters usually have a rated pore size of 0.2 µm or smaller.18 Use of redundant sterilizing filters should be considered in many cases. Whatever filter or combination of filters is used, validation should include microbiological challenges to simulate worst-case production conditions for the material to be filtered and integrity test results of the filters used for the study. Product bioburden should be evaluated when selecting a suitable challenge microorganism to assess which microorganism represents the worst-case challenge to the filter. The microorganism Brevundimonas diminuta (ATCC 19146) when properly grown, harvested and used, is a common challenge microorganism for 0.2 µm rated filters because of its small size (0.3 µm mean diameter). The manufacturing process controls should be designed to minimize the bioburden of the unfiltered product. Bioburden of unsterilized bulk solutions should be determined to trend the characteristics of potentially contaminating organisms”.
Ahora bien, en un artículo publicado en PharmaTech de
Sep 21, 2011, escrito por Erik Greb de Equipment and Processing Report, encontré esta información que traduzco con mis limitaciones para dejar clara la fuente y los reconocimientos correspondientes:
“A medida que el mercado de productos biofarmacéuticos crece, los fabricantes deben estar seguros de que entienden las directrices reglamentarias para la producción de estas terapias. Debido a que la mayoría de los productos biofarmacéuticos son inyectables, su esterilidad es crucial para garantizar la seguridad de los pacientes. La filtración es una estrategia central de aseguramiento de la calidad de los medicamentos inyectables. En una guía de 2004, la FDA dice que el "uso de filtros de esterilización redundantes debe ser considerado en muchos casos" (1). Pero no todos los fabricantes están de acuerdo en lo que es la filtración redundante.
EL informe técnico publicado por la Parenteral Drug Association (PDA) proporciona cierta claridad. El informe define la filtración redundante como "un tipo de filtración en serie en el que un segundo filtro de esterilización se utiliza como respaldo en caso de un fallo de integridad del filtro de esterilización primaria"(2). El segundo filtro de esterilización está incorporado en línea, usualmente antes del tanque de almacenamiento o posiblemente después de éste, para proporcionar seguridad adicional de filtración esterilizante, dice Jerry Martin, vicepresidente senior de asuntos científicos mundiales de Pall Life Sciences, uno de los autores del Reporte Técnico de la PDA en esa época y una gran persona y profesional que hoy está retirado y es un excelente asesor.
"Este enfoque es especialmente prudente cuando el filtro final de esterilización no es probado previamente y los lotes se envasan asépticamente en recipientes pequeños antes de la prueba de integridad del filtro después del uso", dice Martin. "Tales lotes a menudo no pueden ser reelaborados en el caso de una falla de integridad del filtro de esterilización, por lo que la inclusión de un filtro de esterilización redundante puede salvar el lote en el caso de que un filtro no cumpla con la prueba de integridad”.
Algunos profesionales de la industria, utilizan el término redundancia para describir dos filtros, uno de los cuales es terminal, que están separados por un tanque de retención, pero por lo demás muy cerca de un tren de llenado. "Preferimos la definición más estricta de dos filtros usados en serie para cumplir con los objetivos de una sola etapa de procesamiento", dice Rick Friedman, director asociado de la Oficina de Fabricación y Calidad de Producto de la FDA. "Pero, en todos los casos, el aspecto clave es la mitigación del riesgo que ofrece el uso de dos filtros esterilizadores en lugar de uno".
Al parecer la postura de la FDA es asegurar la esterilidad independientemente de la postura de algunos profesionales y la definición se apega a la configuración 4 y menciona doble filtración y no redundancia, reflexión de su servidor.
Teóricamente, la filtración redundante no es necesaria para lograr un producto en llenado aséptico sea estéril, pero algunos farmacéuticos encuentran que mejora la calidad del producto. La incorporación de dos filtros de 0.1 μm en un proceso ayuda a la esterilización de los medios de cultivo celular de forma exitosa, de acuerdo con un profesional de fabricación en una importante empresa biofarmacéutica. "Realmente no llegamos a entender el mecanismo por el cual dos filtros de 0.1 μm son mejores que uno, pero el resultado parece estar diciéndonos que son mejores. Redujo significativamente nuestra taza de contaminación de cultivos celulares ", dice el profesional que prefirió permanecer anónimo.
Otros consideran que la filtración redundante es un desperdicio. "El costo está dentro del análisis del usuario final y, por lo tanto, la filtración redundante debe ser revisada críticamente", dice Maik Jornitz, vicepresidente senior de marketing de bioprocesos en Sartorius-Stedim Norteamérica (en ese momento). Además de aumentar los costos del filtro, la filtración redundante puede obligar al usuario final a abordar las preocupaciones sobre el volumen de retención, la adsorción inespecífica y los lixiviables. Si las funciones de dos filtros no están definidas apropiadamente, un fabricante debe realizar pruebas de integridad en ambos, y no puede utilizar uno de ellos como seguro en el caso de que el otro falle. Las empresas deben preguntarse si la filtración redundante es verdaderamente necesaria, si la estrategia se promulga para complacer a los reguladores, o si es "un soporte de ayuda para un problema más grave", dice Jornitz.
Los fabricantes pueden encontrar que la filtración redundante tiene más valor para algunos procesos que para otros. "La necesidad de filtración redundante depende del proceso y debe basarse en una evaluación de riesgos completamente documentada que incluya la carga biológica de los componentes, la validación de retención de filtros, el historial de integridad del filtro, el tiempo de procesamiento, el valor del producto y el potencial de reprocesamiento", comenta Willkins que es gerente de equipo técnico norteamericano de consultoría para EMD Millipore. "Se recomienda un exhaustivo análisis de costo-beneficio para cada aplicación específica".
Las regulaciones actuales mencionan que muchos casos requieren filtración redundante, aunque la guía sugiere que los fabricantes lo consideran. "El requisito es la comprensión y el control de la biocarga, por cualquier medio que se demuestre que sea robusto y validado", dice Wilkins. Por lo tanto, los farmacéuticos son libres de utilizar su propia discreción sobre la necesidad de esta estrategia, incluso cuando están obligados a demostrar que sus procesos garantizan la seguridad microbiana de sus productos”.
Esta información me dio una luz para este tema, por todo lo descrito es obvio que la tendencia desde algunos ayeres es el aseguramiento total de la esterilidad y, para ello, en procesos asépticos es imperativo cuidar todos los aspectos desde reducción y eliminación de Biocarga de los componentes y materiales, el hecho de asegurar que gases, agua, venteos y todo aquello que pueda estar en contacto con el producto esté perfectamente controlado.
Es importante considerar el aspecto que para tener una estrategia de control de la contaminación y de mitigación es necesario una Gestión de Riesgos de Calidad efectiva con un conocimiento real y práctico de los riegos existentes en todas las etapas del proceso aséptico, usando las herramientas necesarias de identificación, valoración y causa raíz en el entorno del proceso, al parecer la mejor seguridad al carecer del “over kill” y el SAL en los sistemas de filtración es la redundancia, pero veamos qué pasa en México.
Lecturas recomendadas:
- FDA, Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practices (Rockville, MD, Sept. 2004).
- PDA, Technical Report No. 26, “Sterilizing Filtration of Liquids,” Revised 2008, PDA J. Pharm. Sci. Technology. 62 (S-5), 5 (2008)
- ISO-13408-2:2018; Procesamiento Aséptico, Filtración Esterilizante.
- Annex 1; Manufacture of Sterile Medicinal Products; Brussels, 22.8.2022, C(2022) 5938 final
- NOM-059-SSA1-2015
- PharmaTech de Sep 21, 2011, Erik Greb de Equipment and Processing Report
- Handbook of Validation in Pharmaceutical Processes; J. Agalloco, P. Santis, A. Grill, A. Pavell, 2022, CRC press.
Por: Héctor Hugo Téllez Cansino - Facilitador del Conocimiento Técnico Biopharmaceutical System©