Primera parte
Revisaremos los parámetros y las situaciones para realizar la validación de limpieza en equipos que son multiusos y que están en contacto con diferentes activos en la industria farmacéutica. Nuestra revisión es muy general ya que podría ser un tratado de tema de doctorado.
Desde la publicación de la Guía para la Inspección de Validación del Proceso de Limpieza (FDA, 1993), la validación de la limpieza ha recibido una atención creciente por parte de la industria farmacéutica y de las autoridades sanitarias, no quedando circunscrita sólo a la empresa farmacéutica, sino que también se requiere para la industria biosanitaria y alimentaria.
La limpieza es un paso fundamental y obligatorio de cualquier proceso farmacéutico que garantiza la calidad de los productos fabricados. Cada empresa debe optimizar sus procesos de limpieza a nivel de concepción y aplicación, teniendo en cuenta que todo proceso de limpieza no controlado aumenta el precio de fabricación y disminuye su calidad y seguridad.
El proceso de limpieza reduce los residuos, lo cual no significa que los elimine totalmente. Así pues, lo que se busca es disminuir el contenido de residuos hasta un límite preestablecido y aceptable. Una parte fundamental del proceso de limpieza es definir cual es este nivel mínimo aceptable que no supone ningún riesgo para el consumidor final.
La limpieza busca la reducción del contenido de residuos de la superficie de los equipos, de las zonas en exposición y comunes entre los diferentes productos y que son, por tanto, potenciales vectores de transmisión de los residuos de un producto a otro. Esta reducción de residuos debe caracterizarse por ser constante, no aleatoria, y darse una forma repetitiva y reproducible.
El diseño de un proceso de validación implica evaluar un determinado proceso de limpieza, de manera que éste queda absolutamente definido en cuanto a los parámetros críticos del mismo, tales como: tiempo, temperatura, tipo de ciclos, números de ciclos, detergentes, etc.
A causa de los estudios que implica, la validación de la limpieza lleva a una mejor comprensión del proceso, que a su vez permite un mejor control y, como consecuencia, mayor reproducibilidad y eficiencia.
La validación de la limpieza se requiere en el campo farmacéutico para evitar posibles interacciones sinérgicas clínicamente significativas entre productos químicos farmacológicamente activos.
De las causas que han llevado a un mayor control de la contaminación cruzada podemos mencionar la existencia de una nueva generación de productos muy potentes, las trágicas consecuencias de las contaminaciones ocurridas repetidamente como, por ejemplo, con sulfanilamida en más de 100 personas y el hecho de tener en cuenta que existen individuos alérgicos a determinadas sustancias, como es el caso de la penicilina y las cefalosporinas. No olvidando activos altamente contaminantes como los oncológicos.
En la validación de la limpieza se trata de conseguir pruebas documentadas con un alto grado de garantía de que se ha limpiado un sistema o una pieza de un equipo hasta los límites aceptables y predeterminados. Todo esto se realiza para verificar la eficacia de los procedimientos de limpieza y asegurar que no existen riesgos asociados con la contaminación cruzada de los ingredientes activos, detergentes o desinfectantes cuando se elaboran en un mismo equipo.
El programa de validación de la limpieza implica identificar los residuos*, seleccionar el método (o métodos) para su identificación, elegir el método de muestreo, determinar el límite de aceptación de residuos en las superficies de los equipos, calcular el factor de recuperación, escribir el procedimiento y formar al personal implicado. El procedimiento de limpieza se debe repetir, por lo menos, tres veces para demostrar su eficacia antes de utilizarlo en rutina en el proceso de fabricación, así se puede garantizar la validez del estado de “limpio” de los equipos de fabricación durante un periodo de tiempo estudiado.
*Restos de principios activos y sus productos de degradación, excipientes y sus productos de degradación, partículas y polvo ambiental, residuos provenientes de equipos y, finalmente, microorganismos y endotoxinas.
En el planteamiento tradicional, cada principio activo requiere una limpieza diferente y probablemente una validación de limpieza diferente. La novedad de la aplicación de un plan de validación de procesos de limpieza en planta son con base en la gestión de riesgos para disponer de un sistema de gestión de la validación de limpieza para todos los equipos y principios activos utilizados en la fabricación.
A causa de la variedad de los principios activos que podrán entrar en contacto con los equipos, se pretende que cada vez que se utilice un principio activo por primera vez, se introduzca en una matriz de riesgo. Si el riesgo calculado resultante es mayor que el peor caso validado, se debe volver a validar el proceso de limpieza, mientras que si es igual o menor, el principio activo queda incluido dentro del plan de validación establecido previamente y, por lo tanto, no se requiere su re-validación.
Guía para Validación del Proceso de Limpieza (FDA, 1993), particularmente destacaba los siguientes puntos:
- Debe haber procedimientos escritos que detallen los:
- Procesos de limpieza.
- Procedimientos de muestreo.
- Métodos analíticos utilizados incluyendo la sensibilidad de los mismos.
- Los estudios de validación de limpieza deben llevarse a cabo con base en protocolos aprobados y los resultados de los estudios deben documentarse.
- Los procedimientos generales de validación deben mencionar quién es el responsable de realizar y aprobar el estudio de validación, los criterios de aceptación y cuando será necesaria la revalidación.
- Debe haber un informe de validación final, aprobado por el responsable y donde se especifique la gestión que indica si el proceso de limpieza es validado o no.
- Los datos deben incluir la conclusión de que los residuos se han reducido a un nivel aceptable.
Actualmente hay tendencia a reducir al mínimo la intervención humana durante la limpieza para superar la falta de reproducibilidad de la limpieza manual.
En la industria farmacéutica se utilizan tres tipos principales de limpieza: manual, semiautomática y totalmente automática, contando cada uno con sus ventajas y desventajas.
- Limpieza Manual
Este tipo de limpieza se puede definir como la aplicación de una acción mecánica por parte de un operario que usa herramientas y productos de limpieza para limpiar una superficie o equipo. El resultado obtenido después de la limpieza depende directamente de la correcta aplicación y el seguimiento estricto de los procedimientos de limpieza establecidos. El ajuste de los parámetros de control (presión, concentración, temperatura, tiempo, etc.) es responsabilidad exclusiva del operario, por tanto, depende mucho de las habilidades del operario y su formación. La principal ventaja de este tipo de limpieza se dirige a las áreas críticas del material que son de difícil acceso con otros tipos de limpieza. El principal inconveniente es la falta de reproducibilidad del método.
- Limpieza semiautomática
Este tipo de limpieza se puede definir como la secuencia de operaciones tanto manuales como automáticas. La limpieza se efectúa sin desmontar el equipo y la intervención del personal es pequeña, pero importante para que funcione correctamente. Este tipo de limpieza se puede utilizar para equipos que no se pueden desmontar o desplazar.
- Limpieza automática
Este tipo de limpieza no requiere la intervención humana. Está totalmente automatizada. Muy a menudo este tipo de limpieza no requiere un previo desmontaje de los equipos. Esto se logra ya sea por aspersión o por el movimiento de fluidos o disolventes. La secuencia de las operaciones se lleva a cabo bajo condiciones predeterminadas, lo que asegura la reproducibilidad de la limpieza.
Aunque la intervención del operario se reduce al mínimo, es esencial supervisar el buen funcionamiento del sistema con el control de registros secuenciales. Existen dos tipos de limpieza automática: CIP (Clean In Place o limpieza in situ) y COP (Clean Out Place o limpieza fuera de lugar).
- CIP
El sistema de limpieza in situ es un procedimiento de lavado automático que utiliza soluciones químicas de limpieza recirculadas por bombas de alta presión. Esta opción ofrece importantes ventajas para las instalaciones de fabricación, permite una limpieza eficaz y fiable de forma rápida y segura, mejorando la calidad del producto y el cumplimiento de las normativas, sin necesidad de desmontar todo o partes de un equipo. El desarrollo y el diseño de un CIP es especialmente adecuado para equipos donde se fabrican líquidos o semisólidos. Este tipo de instalaciones de limpieza, sin embargo, son costosas y pueden no ser adecuadas para todos los equipos de producción.
Consiste básicamente en ciclos consecutivos de lavados utilizando limpiadores alcalinos, ácidos o intermedios, desinfectantes, seguido de un enjuagado con agua purificada.
Existen dos tipos de sistemas CIP:
- Sistema simple: la solución de limpieza se introduce en el sistema, terminado el proceso se descarga y, por último, se enjuaga.
- Sistema de recirculación: implica la preparación de la solución de limpieza en un tanque externo, debido a que la solución recircula hasta que los ciclos de limpieza hayan finalizado.
Procesos de limpieza
En la tabla se listan los pasos a seguir para llevar a cabo los dos tipos de limpieza: manual y automática o CIP.
Tabla: Etapas de procesos de limpieza.

*Agua del potable de red urbana sin tratamiento en el laboratorio. En todos los casos debe demostrarse que el procedimiento de limpieza es eficaz, consistente y reproducible, con lo cual será necesario validarlo.
La FDA (marzo, 1998) recomienda el uso de CIP para limpiar equipos y tanques de almacenamiento con el fin de reproducir exactamente el mismo procedimiento cada vez. La limpieza manual es más variable que los procesos automatizados por lo que se deben incluir más detalles al escribir los PNO y realizar una rigurosa formación del personal.
Los mecanismos de limpieza
En las operaciones de limpieza, el resultado final depende de cuatro factores interrelacionados.
1- Acción mecánica: se refiere a la eliminación de residuos y contaminantes a través de acciones físicas como el cepillado, lavado y el uso de agua a presión.
2- Acción química: la acción del agente de limpieza. La elección del método y del producto a utilizar depende de la naturaleza de la suciedad y la superficie a limpiar. Son reacciones químicas de oxidación e hidrolisis que rompen los residuos orgánicos y los contaminantes para que sean fácilmente extraíbles de las superficies.
3- Temperatura del agua: desempeña un papel importante en la eficacia de los detergentes porque maximiza su eficacia, por eso se tiene que respetar la temperatura recomendada por el fabricante.
4- Tiempo de acción o de contacto: es el tiempo requerido para que el detergente reaccione con la suciedad con el objetivo de eliminarla.
- Si uno de los factores se reduce, se debe compensar mediante el aumento de uno o más de los otros factores.
- Acción mecánica, como el frotado, cepillado.
- Tiempo de contacto.
- Temperatura
- Acción química, jabón, detergente.
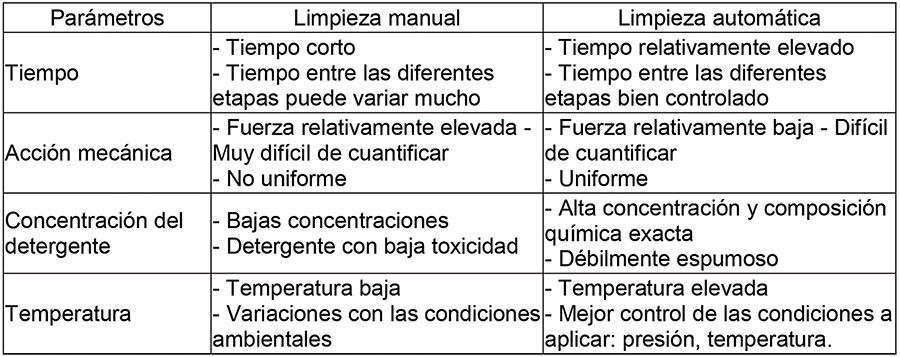
Estos factores intervienen de forma diferente en caso de tratarse de un procedimiento manual o automático como se ve en la tabla siguiente.
Tabla 2: Comparación de los diferentes tipos de limpieza.
Tipos de métodos de limpieza según la frecuencia del lavado
Se pueden definir tres tipos y su implicación para la validación será diferente:
- Métodos de limpieza radical o no serial: elimina residuos de forma exhaustiva → se deben validar de forma completa.
- Métodos de limpieza ordinarios, seriales o parciales: utilizados típicamente tras la fabricación consecutiva de lotes del mismo producto→ no se validan o son objeto de una validación reducida.
- Los métodos de limpieza de repaso o tras inactividad (periodo superior al periodo de validez) →validación reducida centrada en el control microbiológico.
Tipos de validación
Hay tres tipos principales de validación:
1- Validación prospectiva
- Prospectiva. – Concurrente.
- Retrospectiva o reiterada (revalidación).
La validación prospectiva se aplica antes de la distribución de un producto nuevo, o producto elaborado bajo un proceso de fabricación validado. Se hace antes de una fabricación industrial convencional. Se lleva a cabo durante la etapa de desarrollo y es el resultado de un análisis de riesgo en el proceso productivo.
También puede realizarse cuando se prevé efectuar cambios en el proceso de fabricación que pueden afectar a las características del producto.
Según las GMPS y ICH Q7, la validación prospectiva necesita, en general, tres lotes consecutivos con resultados aceptables (aplica también a la validación concurrente) y en algunos casos se puede necesitar lotes o repeticiones de proceso adicionales (por ejemplo, para procesos complejos o extensos).
2- Validación concurrente
La validación concurrente es la forma de validación que se lleva a cabo durante la producción normal. El producto es validado mientras está en producción.
Esta validación es excepcional y sólo se hace con la decisión justificada, documentada, aprobada del personal autorizado (ICH Q7). Requiere aprobación previa por autoridades sanitarias.
La validación concurrente puede efectuarse:
- Cuando todos los datos y parámetros de producción no están disponibles debido a que se produce un número limitado de lotes.
- Cuando los lotes de los principios activos o fármacos se producen con poca frecuencia, o cuando el ingrediente activo es producido a partir de un proceso validado pero modificado.
3- Validación retrospectiva o reiterada (revalidación)
La validación retrospectiva consiste en establecer una evidencia documentada de la idoneidad de un producto o proceso basándose en la evaluación de los datos históricos acumulados existentes.
Esta validación se realiza cuando los procesos se utilizan sin cambios significativos y cuando se dispone de los resultados del control de proceso y del producto final y de limpieza.
La validación retrospectiva se basará en datos históricos. Sus fases incluirán la preparación de un protocolo de validación específico y el registro de los resultados de la revisión de los datos, de los que se extraerá una conclusión y una recomendación.
No obstante es muy difícil hacer esta validación para un procedimiento de limpieza ya que no se analizan residuos indicadores como serían los detergentes o productos de degradación.
Hasta aquí vamos a considerar la primera etapa de la validación de limpieza, en el siguiente artículo hablaremos sobre la documentación y metodología.