
Desde hace unos años, hemos conversado con diferentes actores en la manufactura estéril de diferentes regiones, la importancia que ha tenido la revisión del Anexo 1 de la agencia europea desde 2015 y la larga espera de su autorización por estas autoridades, así como esperar el impacto regulatorio, técnico y en la realidad práctica, así como un tanto de no tener conocimiento del plan de aseguramiento de esterilidad en nuestra región de la USP y su correlación con la estrategia dve contaminación planteado por el anexo 1, han hecho que nos decidiéramos a escribir esta disertación.
Desde el año 2021, la Agencia Europea de Medicamentos (EMA) aprobó la nueva versión del Anexo 1 para la fabricación de productos estériles; esta aprobación después de una discusión exhaustiva en el mundo, desde 2015, impactó de manera importante el estado de regulación de las Buenas Prácticas de Fabricación a nivel global, la respuesta inmediata a esta aprobación fueron las actualizaciones de la Organización Mundial de la Salud (OMS) y el Comité de Inspección Farmacéutica (PIC/S) a los anexos respectivos, México es miembro del PIC/S desde enero 2018, teniendo como deber la implementación de este documento dentro de sus estándares de inspección; esta situación ha colocado a la autoridad mexicana en una posición de tener que revisar y actualizar las normatividad correspondiente y la FEUM para integrar y armonizar los nuevos requerimientos de los mencionados anexos.
Es importante mencionar que el anexo de PIC/S y el de EMA son idénticos en contenido, formas y requerimientos, así como señalar que la FDA es miembro activo de PIC, pero hay que aclarar que, hasta el día de hoy, no hay una posición oficial en cuanto a la observancia por parte de esta agencia a estos anexos y se infiere el reconocimiento implícito, así mismo es importante mencionar que la “Guía para la Industria” correspondiente al Procesamiento Aséptico en Estados Unidos no ha tenido actualización desde 2004 y sigue vigente conforme a lo que aparece en el sitio web de la FDA.
Esta información es importante para comprender el marco referencial para desarrollar la estrategia de contaminación como un plan documental que va a ser revisado en inspecciones por parte de agencias regulatorias.
La definición de CCS conforme a los anexos referenciados es la siguiente:
"Estrategia de Control de la Contaminación (CCS): Un conjunto planificado de acciones derivadas del conocimiento actual del producto y del proceso para mantener los microorganismos, pirógenos y partículas en control, que asegura el desempeño del proceso y la calidad del producto. Los controles pueden incluir parámetros y atributos relacionados con sustancia relacionadas o componentes de sustancias activas, excipientes y productos farmacéuticos, condiciones operativas de instalaciones y equipos, controles de proceso, especificaciones de productos terminados y los métodos asociados y la frecuencia de seguimiento y control".
Este conjunto de acciones para prevenir y controlar la contaminación por microorganismos, pirógenos y partículas en cumplimento, que asegura el desempeño del proceso y la calidad del producto, está vinculada lógicamente con la esterilidad del producto, la representación más clara de libre de contaminación microbiológica es la esterilidad, en Estados Unidos en 2011 la Farmacopea de este país (USP) presentó un capítulo referente a este titulado, “Garantía de Esterilidad”, que menciona textualmente:
“Este capítulo ‘informativo’ proporciona información general sobre los conceptos y principios involucrados en la preparación de materiales que deben ser estériles. Según la definición más estricta de esterilidad, un artículo se considera estéril solamente cuando no contiene microorganismos viables. Sin embargo, no es posible aplicar esta definición textual en artículos reales cuya etiqueta indica que son estériles debido a limitaciones irresolubles en las pruebas. No se puede demostrar la esterilidad sin someter cada unidad estéril a pruebas destructivas. En realidad, la seguridad microbiológica se logra por medio de la implementación de controles interrelacionados que, en combinación, proporcionan la confianza de que los artículos son aptos para el uso indicado en la etiqueta. Los controles son los que proveen la garantía deseada sobre el riesgo microbiológico en vez de los resultados de cualquier prueba en artículos terminados o intermedios del proceso. La verificación de la seguridad de los productos cuya etiqueta indica que son estériles es generalmente conocida como “garantía de esterilidad”
Este texto introduce de manera claramente similar al CCS a la creación de controles interrelacionados, estos integrados por medio de un plan de aseguramiento de la esterilidad (PAE), la FDA considera este plan recomendado en la USP como la estrategia de control de calidad fundamental en la fabricación de productos estériles.
Ambas estrategias, El CCS y PAE plantean la revisión fundamentada en Gestión de Riesgos de Calidad de los elementos funcionales que conforman los procesos de manufactura estéril, desde las materias primas y materiales, hasta el producto terminado disponible para su utilización.
El PAE planteado en <1211> de la USP se presenta en la siguiente figura y plantea la interrelación de los elementos del proceso que permiten la fabricación estéril:
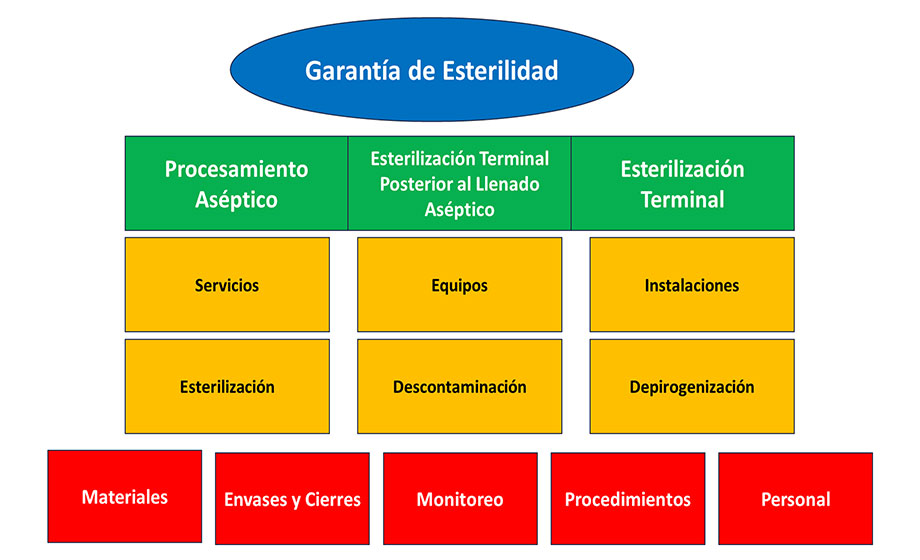
Estos elementos para entender el capítulo de la USP contribuyen en el Aseguramiento de Esterilidad y debe existir un plan de acciones de control para mantener y sostener las variables críticas que garantizan esta. La USP presenta en la siguiente figura los factores que influyen en la producción estéril:
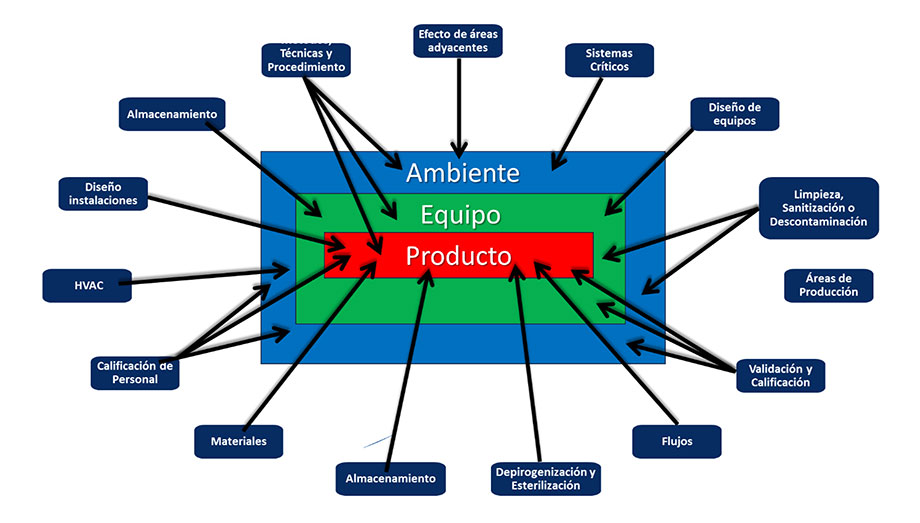
Estos elemento y factores planteados por la USP están perfectamente considerados en la estrategia de control de contaminación, como lo documentó la Asociación de Productos parenterales (PDA) en el Reporte TR# 90 de 2022, bajo el título de “Desarrollo de la Estrategia de Control de Contaminación”, siendo esta asociación gremial de índole global, donde plantea un modelo conforme a esta tendencia regulatoria y representándola en la siguiente figura:
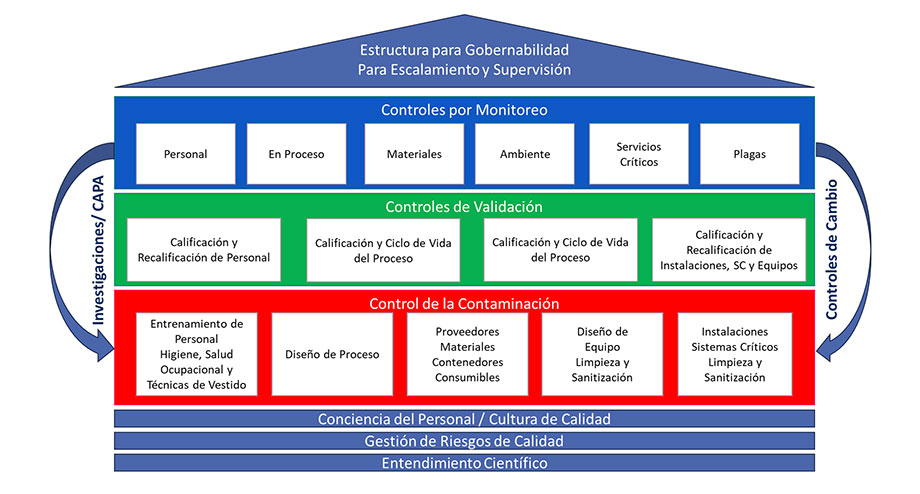
Este modelo establece una estructura para lograr la gobernabilidad del proceso de fabricación por medio de generar tres niveles de control enfocados a la contaminación, validación y monitoreo, fundamentándose en una cultura laboral consiente del impacto de sus actividades en los puntos críticos de control del proceso para prevenir fallas que desvíen o alteren el estado operación, estos puntos determinados por una Gestión de Riesgos efectiva obteniendo las causas raíz de las fallas y transformando el estado basal de proceso en un estado dinámico, activo de monitoreo de puntos clave de desempeño que lleven a el aseguramiento de la calidad y esterilidad del producto.
Paralelamente, es importante decir que la Academia de Buenas Prácticas de Europea creó un documento llamado “Cómo desarrollar y documentar una estrategia de control de la contaminación,– Grupo de Trabajo del TCE sobre Estrategia de Control de la Contaminación – de Enero 2022” que es una base muy útil, para la implementación de estos requerimientos. Este documento plantea un modelo de 3 etapas para la implementación de una estrategia de control de contaminación, estas etapas son:
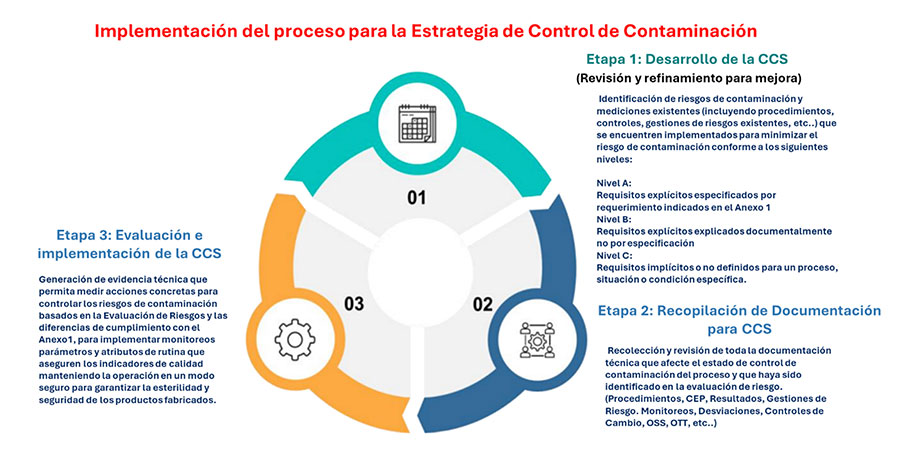
Con toda esta revisión referencial, podemos establecer un modelo que satisface los dos requerimientos tanto el generado por el Anexo 1 y el establecido en el <1211> de la USP, creando un modelo robusto que asegure la esterilidad del producto por medio de la prevención de la contaminación, usando la gestión de riesgos como sistema central de la generación de la estrategia de monitoreo de puntos clave de desempeño del proceso orientados al control preventivo en el sistema estandarizado de calidad.
Este modelo se presenta a continuación:
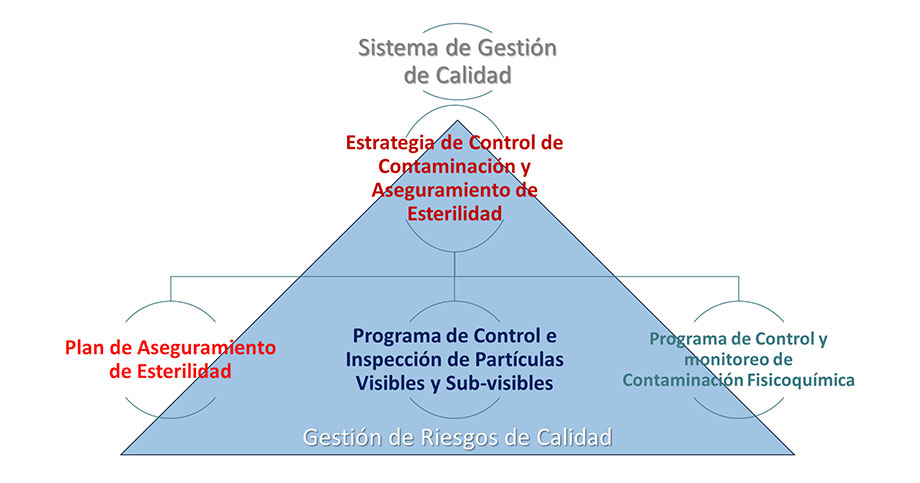
Este modelo, permite:
- Integrar los tres factores clave de los riesgos de contaminación:
- Puntos de vulnerabilidad a la esterilidad relacionados con la biocarga, endotoxina o pirógenos como control de partículas viables.
- Revisar la vulnerabilidad en partículas no viables sin efecto fisicoquímico representadas por partículas visibles y subvisibles.
- Revisión de vulnerabilidad a contaminantes fisicoquímicos como trazas o substancias relacionadas que afecten de forma directa la calidad del producto.
- La estrategia se generaría por medio de la evaluación de peligros de vulnerabilidad y control en los factores claves mencionados, revisando todos los niveles de interacción con los elementos del proceso y haciendo un análisis de brecha de cumplimiento con lo indicado en las normatividades correspondientes.
- Con esta evaluación de riesgos y comprendiendo las causas raíz que generaran las fallas que colocan en peligro de contaminación y riesgo de esterilidad, crear un plan de acción estratégico para incrementar el grado de detección y control en los parámetros y atributos críticos del proceso con los límites de acción y alerta necesarios para mantener el estado de control del sistema de fabricación.
- Este plan de aseguramiento de esterilidad y control de contaminación deberá ser iterativo y revisado continuamente y actualizado anualmente, en caso de cambios o desviaciones que alteren el estado de control del plan, éste deberá ser ajustado inmediatamente para lograr un seguimiento y seguridad de efectividad, usando la gestión de riesgos para este fin, así como el sistema de control de cambios.
- La comunicación continua por medio de fábrica visual permite que todos los dueños y patrocinadores de proceso observen continuamente el mantenimiento del plan de aseguramiento de esterilidad y control de contaminación.
- El control estadístico de este instrumento permitiría que la verificación continua del estado calificado y validado sea adelgazada y efectiva, logrando minimizar pruebas de recalificación y optimando el plan maestro de validación, así como prevendría la generación de desviaciones críticas por descontrol de biocarga y contaminación, y mantendría la seguridad de esterilidad de la fabricación por medio de los indicadores clave.
Esta estrategia integral asegura el estado de cumplimiento actualizado para la manufactura estéril, desde nuestra visión, no existen diferencias substanciales entre ambas formas de estructurar la prevención de la esterilidad y el control de la contaminación ambas son complementarias y la integración permite la generación de un modelo robusto de seguridad operativa.
Integrando todos los elementos y factores que intervienen en el procesamiento de productos estériles de manera total por medio de una Gestión de Riesgos de Calidad efectiva.
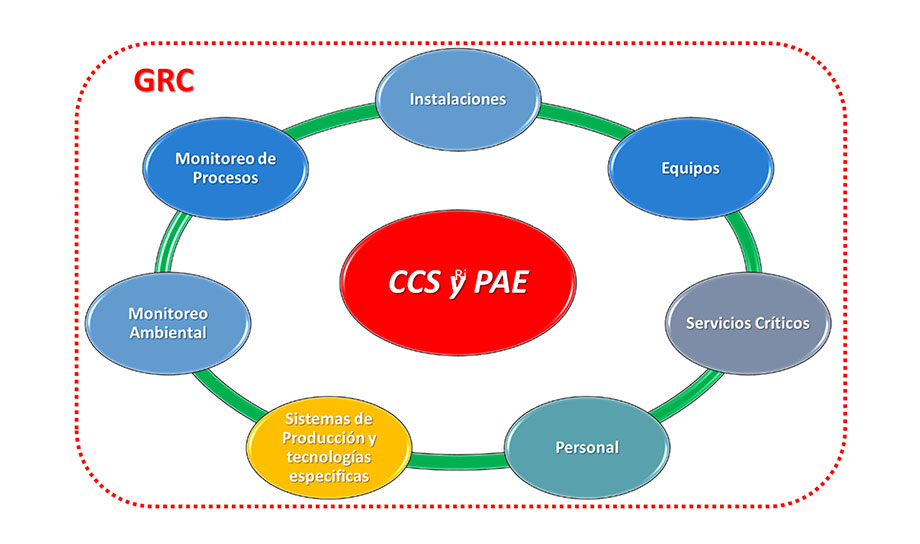
Es desarrollar un plan integral de mantenimiento y sostenimiento de los parámetros críticos de calidad, el detalle de la estrategia dependerán del grado de entendimiento, cultura de calidad y disposición a la mejora de cada empresa, reflejada en sus políticas y procedimientos, esta herramienta de gestión estratégico es la consolidación de la aplicación de la gestión de riesgos de forma integral, hemos tenido la oportunidad de crear el modelo de brechas y análisis de fallas para prevención de peligros en esterilidad y contaminación en fabricación estéril, cada empresa puede seguir este modelo general y robustecerlo conforme a sus necesidades y puedo afirmar que su aplicación es similar a producciones no estériles o aplicaciones diferentes en industrias de ciencias de la vida.
Un punto importante a considerar que, debido a lo reciente de la autorización, no conocemos experiencias reales de implementación de la CCS en la región, así mismo increíblemente es que aunque desde 2011 la USP implemento el capítulo <1211> solamente empresas trasnacionales han generado un plan de aseguramiento de calidad por requerimientos corporativos, la realidad, como sucede con cada tema de vanguardia está empezando un desarrollo de la CCS en la región, ocasionado centralmente por la preocupación de la inclusión del Anexo 1 a las normativas por parte de las autoridades sanitarias.
Adicionalmente, no podemos descartar que efectivamente al ser miembros de PICS y estar tan cerca de la regulación de FDA, las empresas en México que hoy suministran productos de exportación serán obligadas a su implementación y durante ese tiempo seguramente nuestras autoridades sanitarias incluirán esta garantía de esterilidad en la normatividad vigente, luego entonces el reto es aún mayor para México, ya que tendremos que implementarlo en la industria farmacéutica y de también en dispositivos médicos para mantener nuestra competitividad y cumplimiento regulatorio.
En nuestra opinión, esta es la nueva realidad para los sistemas de gestión de calidad, el desarrollo de estrategias fundamentadas en gestión de riesgos proyectivos, pero como dijo el clásico, “hoy eso es otra historia” y en esas tareas de implementación nos encontramos con un nuevo reto, como lo fue en su momento, la gestión de riesgos, los sistemas CAPA, la validación y el inolvidable nacimiento de las buenas prácticas.
Por: Héctor Hugo Téllez Cansigno y Enrique Blanco Vargas*
Referencias:
- USP <1211> Capitulo general “GARANTÍA DE ESTERILIDAD” (2022)
- Annex 1, “Manufacture of Sterile Medicinal Products”, EUROPEAN COMMISSION Brussels, 22.8.2022 versión final.
- PDA, Technical Report # 90, 2022
- “Creating Contamination Control Strategy”, Siegfried Smith, Pharmaceutical Technology, January, 2023.
- How to Develop and Document a Contamination Control Strategy
- – ECA Task Force on Contamination Control Strategy – (2023)
- Annex 1 (Manufacture of sterile medicinal products), PHARMACEUTICAL INSPECTION CONVENTION PHARMACEUTICAL INSPECTION CO-OPERATION SCHEME, PE 009-16 (Annexes), February 2022
- NOM-059-SSA1-2015
*Héctor Hugo Téllez Cansigno y Enrique Blanco Vargas, facilitadores del Conocimiento Técnico. Biopharmaceutical System®







