“Sí hasta los cerros cambian cada año,
que no cambien las cosas”
Dijo el abuelo al nieto...
Reflexión inicial sobre el Control de Cambios
En mi andar por diferentes foros, empresas y conviviendo con diferentes colegas, me han preguntado que desde mi punto de vista cómo aplicaría la gestión de riesgos en los sistemas de control de cambio, dado que continuamente se complica esto o simplemente no se usa en diferentes lugares, increíblemente la “AMEFITIS aguda” o creer que todo se resuelve usando esta herramienta de riesgo ha creado un problema en una evaluación, que es simple, breve y concisa, reflexiono la utilización del método para esta necesidad, recurriendo a la norma de buenas prácticas de mi México, pero el razonamiento es igual para cualquier coordenada geográfica, entonces comencemos.
En la NOM-059-SSA1-2015, se entiende como Control de Cambios a lo siguiente:
3.42 Control de Cambios, a la evaluación y documentación de cualquier cambio que pudiera impactar en la calidad del producto.
Entendiendo esta definición, Control de Cambios es una evaluación técnica documental de eventos, modificación o mejora en los elementos de calidad de las instalaciones, equipos o sistemas relacionados para la fabricación de un producto, que impacta los atributos de calidad establecidos en las bases de diseño del mismo; en otras palabras, es evaluar los impactos en los sistemas de calidad para generar las acciones prioritarias y sincronizadas para realizar este cambio de manera efectiva.
Ahora bien, es importante enfatizar que realizar la evaluación del impacto requiere de una metodología que establezca las herramientas para medirlo, esta metodología es el análisis de riesgo, la norma es clara al pedir el análisis del impacto, esto se hace usando básicamente dos herramientas de gestión de riesgo, el análisis preliminar de daños (PHA) producto de un análisis de brecha (GAP) para conocer las modificaciones al estado inicial y comprender los impactos en el paciente, producto, proceso, personal y patrimonio de la empresa (Regla de las 5Ps), esta metodología no es la gestión completa de riesgo de las posibles fallas asociadas a los elementos modificados, estas probables fallas se estudiarán a detalle cuando se implemente el cambio en dicho los sistemas, equipos, materiales, software o procedimientos impactados, realizando las acciones correspondientes a un ciclo de riesgo completo, estudiando los modos de falla y sus correspondiente investigación de causas para generar las acciones de prevención y corrección que garanticen la funcionalidad y la operabilidad del cambio.
La norma establece para la administración de los Controles de Cambio:
5.7 Control de cambios
5.7.1 Debe existir un sistema documentado de control de cambios que incluya la gestión de riesgos para la evaluación e impacto del cambio propuesto sobre los procesos, proveedores, sistemas críticos, sistemas computacionales, áreas, servicios, equipos, métodos analíticos, especificaciones, documentación, disposiciones regulatorias y calidad del producto.
5.7.2 Los cambios no planeados deben considerarse como desviaciones o no conformidades.
5.7.3 Debe conformarse un Comité o Grupo Técnico integrado por representantes de las áreas involucradas y por el responsable de la Unidad de Calidad, quienes revisarán, evaluarán y aprobarán el cambio propuesto.
5.7.4 Debe darse seguimiento a la implementación de los cambios aprobados y asegurar su cierre de acuerdo a lo previamente establecido.
El racional de este numeral, establece los siguientes puntos clave para esta tarea:
- Sistema documentado que contenga los procedimientos, formatos, registros y controles necesarios para garantizar la aplicación del control de cambios conforme a las buenas prácticas de documentación.
- El uso de una herramienta comprobada para la evaluación de los impactos del cambio y poder establecer las estrategias de control y administración de los riesgos detectados en los procesos, proveedores, sistemas críticos, sistemas computacionales, áreas, servicios, equipos, métodos analíticos, especificaciones, documentación, disposiciones regulatorias y calidad del producto.
- Entender claramente que todo cambio que no está derivado de un proyecto o sea planificado es una desviación al sistema de calidad, una desviación o no conformidad puede crear una actividad o proyecto que genere una modificación al estatus del sistema de calidad y este proyecto o actividad podrá generar un control de cambios, pero un evento que altere las condiciones controladas o estándar del sistema no puede ser un cambio en términos de buenas prácticas.
- Debe existir un mecanismo que asegure la implementación de las actividades generadas para el cambio propuesto, centradas en lo que identifica la evaluación de impactos realizada con foco en riesgo, estas actividades planificadas serán seguidas a través de un cronograma y alertas que aseguren el cumplimiento de fechas y acciones de calidad y regulatorias que garanticen el cumplimiento y puesta en marcha de la modificación garantizando en todo momento el estado de arte de la operación y el mantenimiento de las buenas prácticas de fabricación, este seguimiento debe estar documentado y ser parte esencial para demostrar con las evidencias técnicas irrefutables de la existencia y congruencia de la gestión de los controles de cambio de la organización.
En sí, la gestión de cambios evidencia la capacidad de la organización por medio del sistema de gestión de calidad de adaptarse a las nuevas necesidades, esto para mantener y sostener un proceso de mejora continua confiable y documentado, la razón central para administrar los cambios es garantizar por medio de la evidencia documental que los proceso de fabricación en todas sus etapas, identifican los impactos y las consecuencias de las modificaciones en que los aspectos operativos no alteren la funcionalidad, la calidad y, sobre todo, protegen a los usuario o pacientes en esta actividad natural de las empresas.
Tipos de Cambios
Los cambios que afectan un proceso se pueden dividir en unas pocas categorías principales. Aunque esto ciertamente está sujeto a debate, podríamos aproximarnos a esto de la siguiente manera:
- Cambios materiales. Pueden incluir cambios a los materiales activos y excipientes, sus fuentes/proveedores aprobados, especificaciones, cantidades relativas en una formulación, etc. La mayoría, si no todos, de estos cambios caen en la categoría de Control de Cambios.
- Cambios en el equipo. Pueden aplicarse a cualquier equipo utilizado en las instalaciones de fabricación de GMP. Pueden ser reemplazos exactos, equivalentes funcionales o incluso configuraciones o tecnologías totalmente nuevas. Los cambios de sitio y de ubicación dentro del sitio entran en esta categoría (siempre que no haya cambios que los acompañen, cambios materiales o de procedimiento). Algunos de éstos, pero no todos, caen bajo el Control de Cambios. Cuáles y cómo decidir, se discuten en una sección posterior.
- Cambios de procedimiento. Son cambios en los procedimientos escritos, incluidos los SOP, registros de lotes maestros, formularios de datos, métodos analíticos y otros documentos que se utilizan para respaldar la fabricación GMP. Los cambios en la documentación pueden ser necesarios debido a un cambio de material o equipo, pero también pueden ser necesarios por otras razones, como el cambio de responsabilidad o la ubicación de una operación. La ampliación se superpone a los cambios de procedimiento, de equipo y, a veces, incluso de material.
- Cambios de software. Aunque los cambios de hardware de la computadora se pueden considerar similares a los cambios de equipo, se puede considerar que estos cambios caen en la categoría más amplia de cambio de procedimiento. Esto parece razonable porque ciertos elementos del software, pero no todos, pueden definirse como "registros de lotes" u otros registros relevantes para la fabricación, prueba y lanzamiento del producto. El software como documentación es lo suficientemente diferente de la documentación tradicional como para ser identificado por separado.
Esta categorización es una interpretación, cada organización conforme a su filosofía podrá establecer los tipos de cambio; algunas organizaciones separan los cambios en procedimientos dentro las revisiones históricas o las modificaciones en software dentro de la administración de las unidades de Tecnologías de Información, pero básicamente los cambios en equipos, operaciones de fabricación, implementos, instalaciones o servicios críticos se mantienen como el centro del efecto en la calidad del producto, cabe mencionar también que los cambios en materiales de empaque algunas organizaciones los consideran por separado, pero eso depende del diseño del Sistema de calidad. Desde un punto de vista netamente práctico, esta es la visión de los tipos de cambio.
Utilización de la Gestión de Riesgos en el Control de Cambios
Con varios colegas y en mi andar me han comentado varias formas de utilizar o interpretar la gestión de riesgos en la administración de los controles de cambio, desde mi entender en este tema, la aplicación de la gestión de riesgo a esta actividad es la evaluación de los impactos generados por el cambio propuesto, es decir, cuando se plantea un modificación en el estado actual generando un nuevo estado, existe la probabilidad de un impacto sobre los elementos de calidad del proceso, el siguiente esquema representa este cambio de estatus:
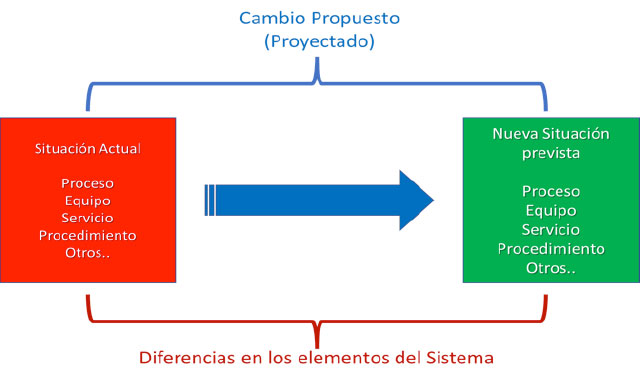
El riesgo que debe evaluarse no se considera basado en la probabilidad de falla del proceso, equipo, servicio, etc.., sino está considerado como una evaluación del impacto en la calidad y su probable efecto en el usuario o paciente como una medida de factibilidad de realizar la modificación a la situación actual a una nueva situación. Esta evaluación pretende generar un conjunto de acciones para garantizar la implementación de forma segura el cambio o nueva situación, en términos prácticos, en mi experiencia, para utilizar de forma sencilla la gestión de riesgo usaría dos herramientas básicas, el análisis de brecha (GAP) y un análisis preliminar de daños modificado (PHA) para evaluar los efectos en los elementos de sistema mencionados en la norma, “sobre los procesos, proveedores, sistemas críticos, sistemas computacionales, áreas, servicios, equipos, métodos analíticos, especificaciones, documentación, disposiciones regulatorias y calidad del producto” y el impacto sobre el paciente, producto, proceso, personal y patrimonio y con esta base generar en los elementos del sistema las acciones a ser planificadas para asegurar la implementación y el cierre del cambio propuesto y su medición.
Para esto, clasificaría los cambios en dos grupos básicos para realizar la primera filtración y lograr administrar de forma básica la criticidad de los cambios a evaluar:
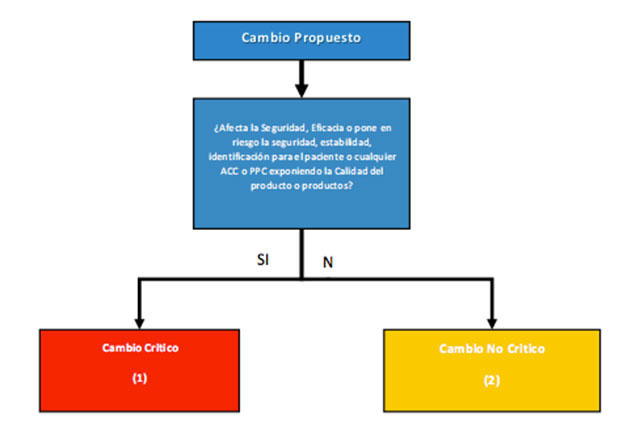
Considerando de forma específica cómo afecta el cambio propuesto la seguridad de uso, la calidad representada por los Atributos de Calidad (ACC), la estabilidad del producto, cómo altera o modifica los Parámetros Críticos de Control, qué asegura el estado validado del proceso como medidas directas del cuidado de la calidad del producto, esta separación permitiría de forma sencilla clasificar los cambios en dos grupos, los críticos sí afecta lo mencionado y los no críticos.
Con lo anterior en mente, podemos clasificar de manera clara qué elementos pueden ser considerados para contener cambios críticos o no críticos, esto lo presento en la siguiente clasificación de tipos de cambios.

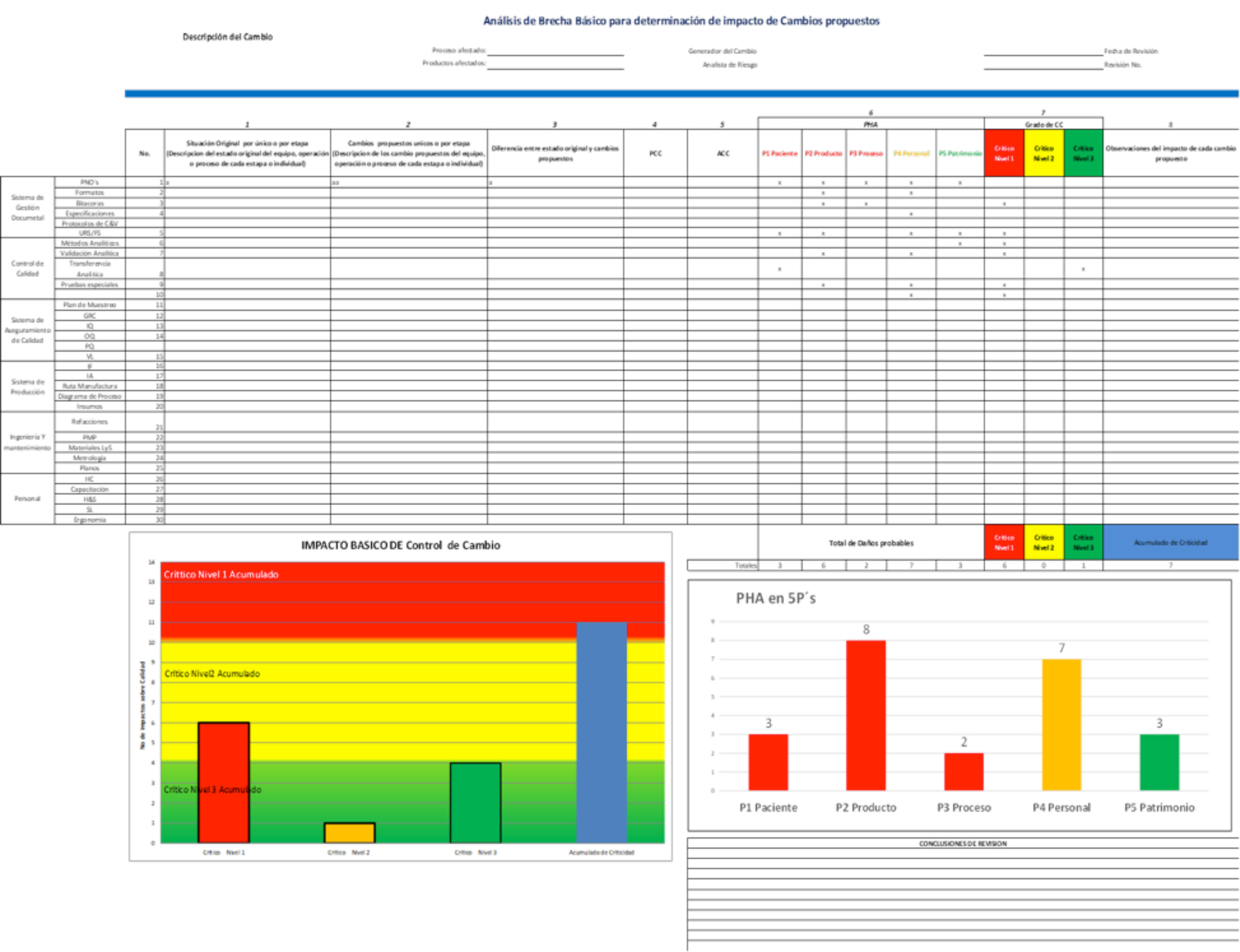
Basándose en esta tipificación de cambios, la evaluación de impactos necesaria podría ser filtrada a través de realizar las siguientes acciones, primero tener una descripción clara y detalla de la propuesta de cambio por cada elemento del sistema de calidad impactado (sistema documental, procedimientos, equipos, áreas, etc.), comparar los cambios en cada elemento contra la situación actual a ser modificada y realizar un análisis de brecha indicando las diferencias entre cada estado, con esta base estimar el impacto en las 5P´s mencionadas y por medio de una tabla de prorrateo valorar el grado de impacto para definir el nivel de criticidad de cada una de las actividades por elemento y totalizar todas y, con ello, establecer el nivel total de criticidad del cambio propuesto para definir las acciones ejecutivas para planificar la implementación del cambio propuesto con sus evidencias técnicas y el cronograma de seguimiento en la figura #1 (al final de este artículo) se presenta la hoja de cálculo para efectuar este análisis de riesgo y tenerlo como base de documentación para el cumplimiento establecido en la norma.
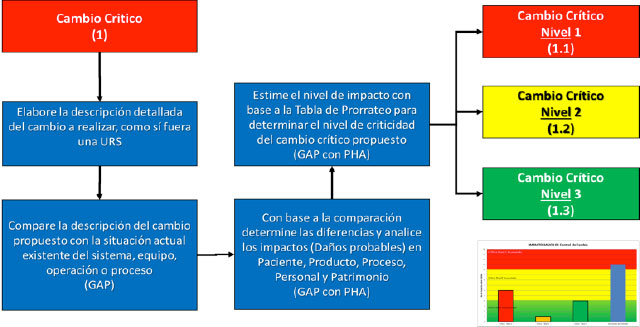
Las acciones recomendadas para los Controles de Cambio, Críticos por los niveles se presentan a continuación:
Cambio Crítico Nivel 1:

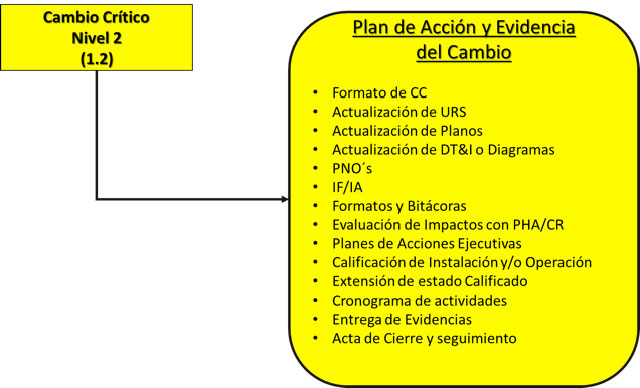
Cambios Críticos Nivel 2:
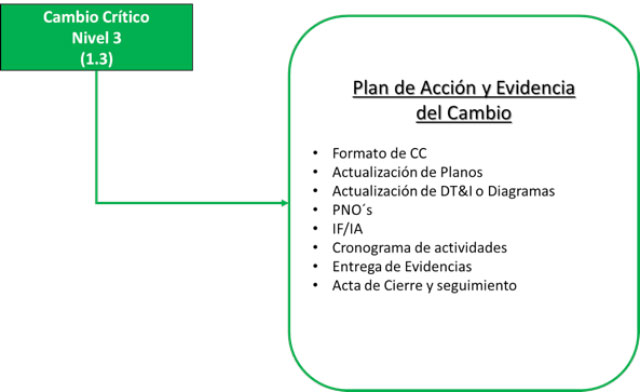
Cambios Críticos Nivel 3:
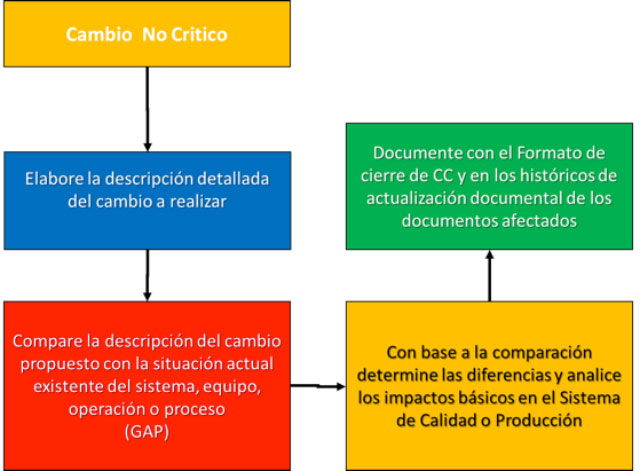
Finalmente, para los cambios no críticos, tendríamos que realizar la descripción detallada, hacer la comparación GAP simple, determinar los impactos, pero sólo documentarlos en los formatos de cierre para asegurar las evidencias correspondientes en conformidad a las buenas prácticas correspondientes.
Conclusiones
La intención de este artículo es proporcionar una idea práctica, obviamente es necesario entrar en detalles para construir de manera clara y en procedimientos esta herramienta, crear los formatos adaptados a su organización y conforme a sus necesidades, lo importante es demostrar la factibilidad y cómo pueden ver el uso del AMFE es enfocado ya en el momento de la implementación para generar CAPA basados en herramientas de causa raíz adecuadas (FTA/ISHK/BT) y entonces calificar, validar o lo que sea necesario para la implementación exitosa de los cambios necesarios.
En otra entrega platicaremos de los cambios llamados igual por igual, igual por equivalente, igual por mejor o igual por alterno, que debido a esto el uso de AMFE sobre los sistemas no sobre el cambio es fundamental.
Figura #1. Ejemplo de hoja para la evaluación de impactos en Control de Cambio usando el método de brecha y la evaluación de la 5P´s:
Por: Héctor Hugo Téllez Cansigno
Facilitador del Conocimiento Técnico Biopharmaceutical System.
Químico Farmacéutico Biólogo, egresado de Universidad Autónoma Metropolitana, plantel Xochimilco, con 40 años de experiencia en la industria farmacéutica en producción, desarrollo, ingeniería, gestión de riesgo, validación, actualmente capacitador y asesor en Biopharmaceutical System con más de 500 cursos y artículos sobre gestión de riesgo, validación, producción, proceso aséptico y más...