Comunicado. La FDA emitió recientemente una alerta sobre las reacciones graves, incluyendo muertes si no se diagnostica y se trata a tiempo, asociados a los medicamentos anticonvulsivos clobazam (conocido como Onfi, Sympazan, y en forma genérica) y levetiracetam (comercializado como Keppra, Keppra XR, Elepsia XR, Spritam, y también en forma genérica).
Se trata de una reacción de sensibilidad a fármacos con esosinofilia y síntomas sistémicos (Dress, por sus siglas en inglés), que puede comenzar como una erupción en el cuerpo.
Esta grave sensibilidad es una reacción del sistema inmunitario que puede causar hinchazón (inflamación) grave en todo el cuerpo o lesiones en órganos como el hígado, los riñones, los pulmones, el corazón o el páncreas. Puede conducir a la hospitalización y al daño o fallo de estos órganos, y puede progresar hasta la muerte, especialmente si se retrasa el tratamiento.
La agencia regulatoria internacional ha exigido a los fabricantes de estos medicamentos adicionar nuevas advertencias sobre el riesgo de Dress a la información de prescripción y la guía para pacientes.
En Panamá, la Dirección Nacional de Farmacia y Drogas del Ministerio de Salud (MINSA) tiene registrados productos con el principio activo levetiracetam, un medicamento anticonvulsivo aprobado para controlar ciertos tipos de convulsiones en monoterapia o con otros medicamentos en niños o adultos como convulsiones mioclónicas o convulsiones tónico-clónicas.
La directora Nacional de Farmacia y Drogas del Minsa, Elvia Lau, explicó que el Centro Nacional de Farmacovigilancia tiene registrados cinco reportes de sospecha de reacción adversa relacionada con el principio activo levetiracetam. Además, precisó que las reacciones adversas relacionadas con el fármaco que han presentado las personas son dolor muscular, falta de apetito y trastornos psiquiátricos.
Por ello, la Dirección Nacional de Farmacia y Drogas del Minsa emitió el pasado 5 de diciembre una nota de seguridad sobre estos dos fármacos.

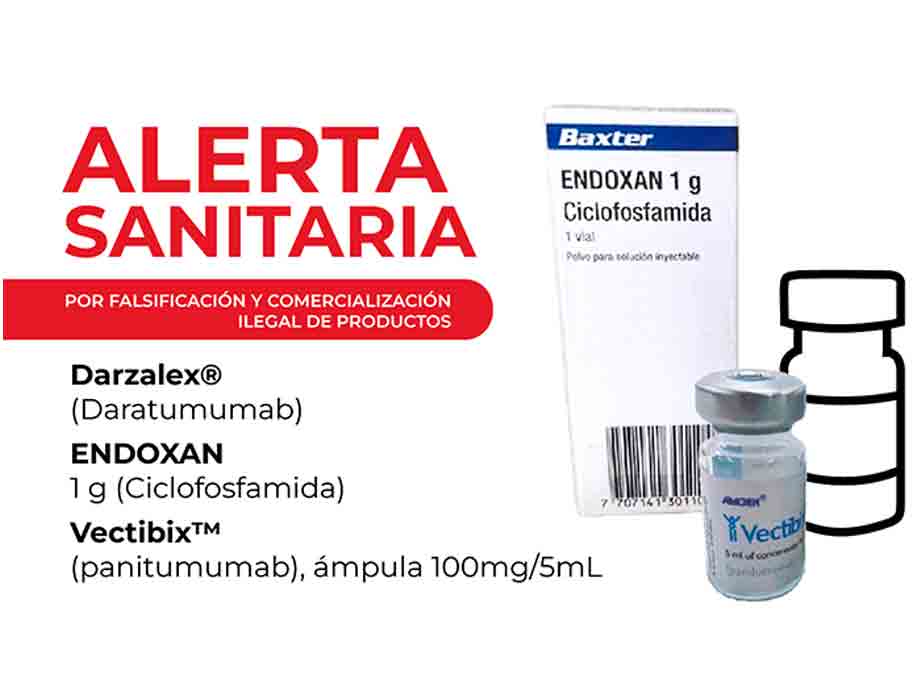
Hoy también publicamos las siguientes notas y más...
Hi-Tech Pharmaceuticals presenta tratamiento para control del peso
Argentina notifica a la OMS primer caso de en humano del virus de encefalitis equina