Comunicado. En México, el aneurisma aórtico abdominal (AAA) representa un problema de salud pública que, aunque poco conocido, puede ser mortal, al reportarse anualmente más de 175 mil muertes en el mundo por esta causa.
De mayor frecuencia en hombres que en mujeres, principalmente a partir de los 60 años, el AAA es una dilatación anormal en la parte baja de la arteria principal del cuerpo llamada aorta, arteria principal del cuerpo que se extiende desde el corazón hasta el centro del pecho y al abdomen.
“Esto ocurre cuando las paredes de la aorta se debilitan, provocando un ensanchamiento o abultamiento, el cual si no es identificado a tiempo puede romperse, causando una hemorragia interna grave con consecuencias fatales” , expresó Enrique López Ochoa angiólogo y cirujano Vvascular del en el Centro Médico Dr. Ignacio Chávez en Hermosillo, Sonora.
Con frecuencia, los AAA crecen de forma lenta sin presentar síntomas, lo que dificulta su detección. Algunos aneurismas nunca se rompen, otros permanecen pequeños o pueden crecer con el paso del tiempo. Sin embargo - agregó el especialista- algunas manifestaciones que revelan el crecimiento de un aneurisma es la presencia de un dolor constante en el área abdominal, dolencias en la espalda, sensación de un latido cerca del ombligo e incomodidad al caminar.
Ante este panorama, es fundamental aumentar el conocimiento y la concientización de este padecimiento silencioso y oculto, promoviendo su detección temprana a través de estudios como el ultrasonido abdominal, especialmente en personas con factores de riesgo como hipertensión arterial, tabaquismo, colesterol elevado, obesidad y aquellos con antecedentes familiares, enfatizó López Ochoa.
“Si bien un AAA diagnosticado a tiempo puede ser controlado y vigilado a través del control farmacológico de factores de riesgo (como la hipertensión arterial, las dislipidemias y el abandono de hábito tabáquico), dependiendo del tamaño, la velocidad de crecimiento y los síntomas que presente el paciente se puede evaluar la posibilidad de llevar a cabo un procedimiento quirúrgico con el objetivo de prevenir su rotura, complicación que puede comprometer la vida”, expuso el especialista.
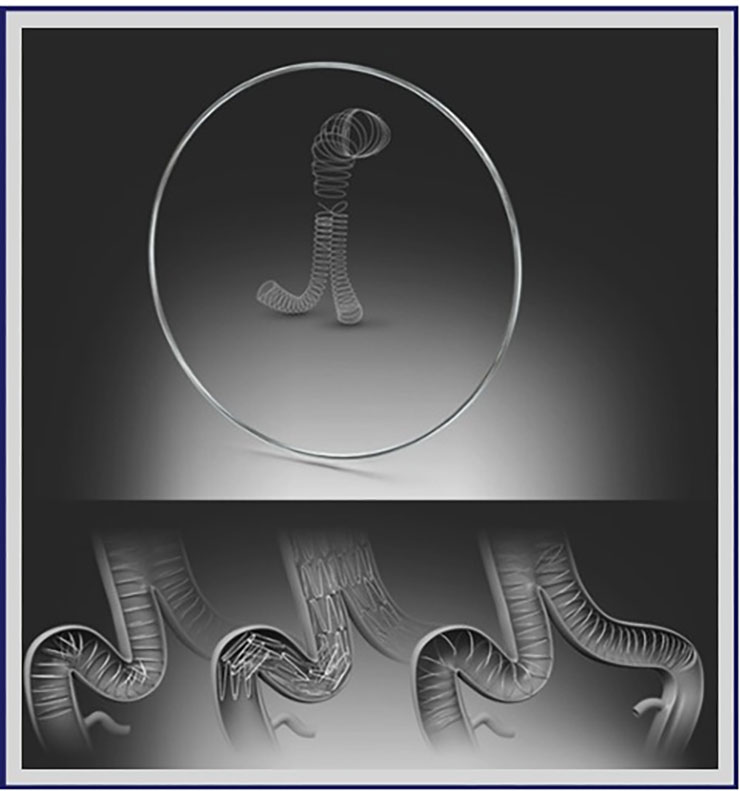
Por fortuna, Sonora se consolida como un referente en atención médica al ofrecer tratamientos innovadores de vanguardia, como un procedimiento quirúrgico denominado Reparación Endovascular de Aneurisma Aórtico Abdominal (EVAR), técnica mínimamente invasiva consistente en la realización de pequeñas incisiones en la ingle para introducir un catéter hasta llegar al aneurisma y cubrirlo con un stent-injerto de alta tecnología, conocido como prótesis Aorfix, capaz de fortalecer la pared arterial, reduciendo el riesgo de ruptura y sangrado, ofreciendo así una solución eficaz y segura para quienes enfrentan esta condición de salud.
La prótesis Aorfix es un dispositivo médico diseñado para el tratamiento de aneurismas de la aorta abdominal, especialmente en pacientes de alto riesgo con anatomías complejas, como aortas tortuosas o con angulaciones, donde otras endoprótesis podrían no adaptarse correctamente, y cuyo diseño flexible minimiza el riesgo de complicaciones durante su colocación, además de promover una recuperación más rápida en los pacientes.

Hoy también publicamos las siguientes notas y más...
GSK continúa impulsando el futuro de la medicina respiratoria
México avanza en 85% de la meta en la Semana Nacional de Vacunación 2025